III. UN REACTOR DE BARRO
EN LAS páginas precedentes hemos descrito numerosos detalles sobre la estructura de las arcillas y los procedimientos que los científicos utilizan para modificarla. Esperamos que el lector se haya convencido de que, desde el punto de vista experimental, no se requiere de técnicas muy complicadas para reorganizar parcialmente la geometría de las arcillas. Cuenta el experimentador con un material de partida que, en su estado natural, presenta ya una estructura muy compleja, la cual ha ido conociendo cada vez con mayor precisión gracias a los avances científicos que poseemos actualmente para caracterizar a los sólidos.

Los químicos, físicos, geólogos y, en general, los estudiosos de lo que hoy en día se conoce como ciencia de los materiales, unen sus conocimientos para convertirse en "maestros de obras". Con picos y palas químicos ensanchan galerías que luego son apuntaladas con nuevas columnas, dando así al entorno una nueva "decoración". Pero los cambios generados son más sutiles y van más allá de la mera expansión del espacio interlaminar, ya que traen consigo modificaciones sustanciales a las propiedades físico-químicas de las arcillas.
Debemos destacar que algunos de estos procesos de modificación se realizan en cierta medida en forma natural y el hombre, desde épocas tan remotas como su propia historia, ha sacado provecho de ellas empleando estos sólidos con diversos propósitos.
Sería presuntuoso de nuestra parte describir todas las aplicaciones potenciales de las arcillas, tanto presentes como futuras. Nuestro enfoque se limitará a describir cómo las propiedades de estos materiales son útiles para promover los procesos de transformación química; es decir su papel como catalizadores.
Hagamos por lo tanto un breve resumen de las propiedades que adquieren las arcillas al introducirse cambios en ellas:
1) Pueden alojar en el interior de su estructura cierta cantidad de agua, dependiendo del tipo de elemento químico presente entre las láminas. Se dice en este caso que el material es hidrofílico.
2) El carácter hidrofílico del material puede ser suprimido parcial o totalmente intercambiando los elementos químicos iniciales por otros compuestos y convertirlo en organofílico, o sea que la arcilla adsorbe moléculas de tipo orgánico.
3) Como consecuencia de las manipulaciones de modificación que se realizan, el mineral acepta o rechaza la penetración de moléculas en su estructura interna en función del tamaño de las mismas, por lo que se comporta como malla molecular.
4) El "obús químico" utilizado para separar las láminas puede a su vez producir pilares que hacen más rígida la estructura original, impidiendo que se colapse como un castillo de naipes.
5) Por último, estas arcillas son sólidos ácidos, comparables a los inorgánicos como el sulfúrico o clorhídrico.
Si consideramos que cada una de las propiedades anteriores puede tener una gama de valores diferentes, es fácil visualizar el número enorme de estructuras con propiedades específicas que pueden ser diseñadas.
Hemos dicho anteriormente que limitaremos la descripción de los usos de las arcillas modificadas al campo de la transformación química, pues pensamos que es allí donde existen perspectivas muy interesantes de empleos útiles para la sociedad, aunque también mostraremos que las arcillas han tenido un muy importante papel protagónico como catalizadores en los procesos biológicos que generaron las primeras moléculas orgánicas en nuestro planeta y, millones de años después, también participaron en la gestación del petróleo.
Desde el punto de vista tecnológico, el empleo de catalizadores en la industria de la transformación ha tenido como propósito fundamental realizar la conversión de los reactivos con un máximo de rendimiento en productos y abatir los costos de producción.
Actualmente se diseñan procesos en los que se trata de invertir la menor cantidad posible de energía y, conocida la problemática que existe alrededor de la degradación ecológica de nuestro planeta, se buscan materiales cada vez más selectivos para orientar la conversión, con mayor precisión, hacia productos más puros, impidiendo las reacciones secundarias indeseables. La madurez e importancia de esta disciplina de la ciencia es tal, que se estima hoy en día que en los países altamente desarrollados el 20% del producto interno bruto se deriva de procesos que dependen de catalizadores. Sin embargo, la tecnología a base de catalizadores tuvo inicios muy peculiares, describiremos algunos que, de paso, nos permitirán subrayar algunas de sus características que los hacen tan importantes en nuestra sociedad.
En 1815, Humphry Davy (1778-1829) descubrió que cuando se introducía un alambre caliente de platino en una mezcla de aire y gas, el metal se volvía incandescente. Davy consideró que se producía oxidación sin que se generara una flama y también notó que, al finalizar la reacción, el metal permanecía aparentemente inalterado. Con esa información, decidió fabricar una lámpara que no reaccionara en forma explosiva al ponerse en contacto con mezclas de gases. El dispositivo en cuestión consistía en una llama protegida por un cilindro que contenía una tela metálica a través de la cual pasaba el oxígeno que la alimentaba. De esta manera el calor era disipado por el metal, y los gases explosivos que pudiera haber fuera de la lámpara no se inflamaban. La lámpara de seguridad de Davy, como se le conoció en su tiempo, se empezó a usar en 1816 en las minas de carbón de Inglaterra, donde se producían numerosas explosiones con las consecuentes pérdidas de vidas, ya que, al aumentar la profundidad de las excavaciones se presentaba el inesperado peligro del gas grisú. Este gas, constituido básicamente de metano (CH4), al liberarse y entrar en contacto con el aire, forma una mezcla explosiva al contacto con una flama. Se cuenta que al llevar la lámpara al interior de las minas, en enero de 1816, y observar su eficiencia, uno de los directores exclamó complacido: "Por fin hemos sometido al monstruo." A Davy esta aportación le valió un título nobiliario en 1818.
Hoy en día sabemos que el platino preoxida en forma parcial a los gases con lo cual, al combustionarse totalmente, no lo hacen en forma explosiva. La acción del platino fue lo que más tarde Berzelius llamaría catálisis. Sin duda la lámpara de Davy marca un hito en las relaciones entre ciencia y tecnología, como también en el uso de esta última con fines humanitarios, más que económicos.
Edmund Davy (1785-1851), primo de Humphry, publicó en 1820 un artículo sobre la preparación de platino finamente dividido que obtenía reduciendo una solución de sulfato de platino por medio del alcohol. El material resultante tenía la propiedad de mostrarse sumamente activo a la temperatura ambiente. Los experimentos de Edmund Davy llamaron la atención de otro químico, esta vez alemán, Johann W. Dobereiner (1780-1849) quien prosiguió los experimentos de Edmund obteniendo un producto que hoy en día conocemos como negro de platino. El material se tornaba incandescente a la temperatura ambiente en presencia de oxígeno, y también lo hacía cuando se le sustituía por hidrógeno. Dobereiner decidió que sus observaciones debían tener una aplicación: inventó un encendedor automático del que se dice se vendieron en su época más de un millón, y que representa una de las primeras aplicaciones de la catálisis en el desarrollo tecnológico.
El aparato consistía en un pequeño compartimiento que, al abrírsele una válvula ponía en contacto una solución diluida de ácido sulfúrico con zinc metálico. De esa manera se llevaba a cabo la siguiente reacción:
El chorro de hidrógeno, producto de la reacción, era transportado a otro recipiente que contenía platino finamente dividido, el cual se prendía al momento. El aparato, a pesar de su éxito inicial no se fabricó por mucho tiempo pues el platino era, por una parte muy caro y, además, se desactivaba rápidamente debido a las impurezas que acarreaba el hidrógeno generado en el primer compartimiento. La acción catalítica del platino habría de cobrar importancia en la química industrial, pues llevaba a cabo ciertas reacciones rápida y fácilmente con su sola presencia. Algunos años después se descubrió un método mejor para la producción de ácido sulfúrico basado en la catálisis del platino.
A finales del siglo XIX se había realizado un gran número de reacciones
en las que se hacía intervenir al platino y otros metales conocidos para convertir
diversas moléculas gaseosas. La oxidación del amoniaco por el platino producía
óxido de nitrógeno y agua, ambos precursores del ácido nítrico, con el cual
se producen fertilizantes. Se conocía además la conversión catalítica del hidrógeno
y el oxígeno para formar agua en presencia del platino y otras reacciones que
en algunos casos tuvieron aplicaciones tecnológicas muy importantes y que dieron
a la catálisis gran prestigio. Sin embargo, el fenómeno catalítico carecía de
una explicación aceptable, a pesar de que se habían avanzado muchas teorías.
Por ejemplo, Michael Faraday explicaba que la conversión del hidrógeno y el
oxígeno se llevaba a cabo sobre la superficie del metal en el cual se condensaban
los dos tipos de moléculas y reaccionaban por su sola proximidad. Hoy en día
sabemos que para que la reacción se lleve a cabo, tanto el hidrógeno como el
oxígeno deben disociarse; desafortunadamente en 1834, fecha en que Faraday publicó
sus observaciones, no se sabía que estas moléculas son diatómicas.
En 1915, Irving Langmuir (1881-1957), un químico estadunidense que trabajaba en la General Electric, propone una teoría sobre la acción catalítica de las superficies metálicas que pone punto final a las especulaciones sobre el fenómeno. El trabajo de Langmuir en sus inicios tenía un propósito más práctico, pues su tarea consistía en tratar de alargar la vida de los filamentos de tungsteno que se empleaban en los focos. Los filamentos se encerraban en una cámara de vacío, pues se suponía que la presencia del aire provocaba la rápida oxidación del metal.
Langmuir demostró que en el vacío, el tungsteno se evapora lentamente, por lo que el hilo se rompe, además de señalar que esta degradación se podía hacer más lenta si se introducía un gas, como el nitrógeno, con el cual el metal no se combinara. Cumplida su misión, Irving decidió estudiar el efecto que producía sobre las superficies metálicas calientes la adición de diversos tipos de gases, llegando a elaborar una teoría que, entre otras cosas, nos dice que la catálisis es un proceso que, en el caso de las moléculas gaseosas que interaccionan con una superficie metálica como condición previa a la reacción, aquellas deben quedar atrapadas en "sitios activos" con los que interaccionan químicamente. La fuerza de dicha interacción es de una magnitud similar a la que hace que los átomos de un sólido mantengan su cohesión. Estas observaciones, entre otras que hizo Langmuir, permitieron a los investigadores interesarse en los estudios sobre las propiedades físicas y químicas de las especies involucradas en el proceso catalítico y tuvieron repercusión profunda en la ciencia de la catálisis, ya que la elucidación del número y naturaleza de los sitios activos en un catalizador, es labor que desde el punto de vista científico y el tecnológico sigue siendo piedra angular para el futuro de esta ciencia. Ya en 1921 Langmuir decía:
| La mayoría de los químicos piensan que la naturaleza de la acción catalítica es tan misteriosa hoy en día como lo era en la época de Faraday, sin embargo con los conocimientos que cada día se van generando sobre la estructura de los cuerpos sólidos y los átomos y moléculas que los componen, deberemos gradualmente tener una visión cada día mejor respecto al mecanismo de acción de dichas superficies. |
LOS SITIOS ACTIVOS EN LAS ARCILLAS
Se ha mencionado anteriormente que una de las propiedades de las arcillas que las hace materiales útiles en la catálisis es su comportamiento como sólidos ácidos.
En el estudio de la química de las soluciones, el investigador es afortunado ya que trata con moléculas de características bien definidas. La interacción entre un ácido, una base y un solvente puede ser, por lo tanto, calculada empleando principios químicos bien probados. Por el contrario, la comprensión de las interacciones ácido-base en los sólidos es muy difícil, ya que la naturaleza de su superficie no está plenamente definida y muchas veces no se cuenta con datos veraces sobre la composición de los complejos que se forman entre el reactivo y el sólido. A pesar de las diferencias entre la química de las soluciones y la de los sólidos, se usan los mismos tipos de conceptos que pasaremos a revisar en forma breve.
Un ácido, tal y como lo describió J. Bronsted (1879-1947) en 1923, es una sustancia capaz de liberar un ion de hidrógeno. Posteriormente, G. N. Lewis (1875-1946) amplió el concepto de acidez superficial y consideró ácido toda aquella molécula provista de un orbital libre que tiende a unirse a una molécula que posee un par de electrones y que funciona como base.
Un ejemplo clásico es la reacción entre el amoniaco (NH3), que es una base, y el hidruro de boro (BH3), que funciona como un ácido:
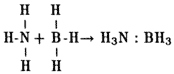
Para Lewis, el ácido más simple es evidentemente el ion de hidrógeno (H+). La concepción de Lewis se basa por tanto en la regla clásica según la cual el octeto constituye la configuración electrónica más estable (dicha configuración es la de los gases nobles, como el helio o el argón, que no reaccionan con otros elementos). Por otra parte, el carácter ácido de una superficie en el sentido de Bronsted depende de la presencia de grupos funcionales susceptibles de liberar iones de hidrógeno.
LOS SITIOS ÁCIDOS EN LAS ARCILLAS
Trataremos ahora de conciliar los conceptos de acidez en las soluciones con la que se presenta en los sólidos, como es el caso de las arcillas, y responder a la pregunta de por qué es importante conocer la naturaleza y fuerza de estos centros. Para ello es necesario revisar brevemente la importancia que representaban estos estudios dentro del contexto de catálisis.
El primer método que se empleó para obtener gasolina a partir del petróleo fue por calentamiento. Cuando aparecieron en el escenario los primeros catalizadores de inmediato resaltó que la naturaleza y cantidad de productos que se obtenían eran diferentes para cada caso, lo que hizo sospechar que el mecanismo de las reacciones era diferente. A fin de poder identificar con precisión los productos de las reacciones se empezó a trabajar con hidrocarburos aislados, haciéndolos reaccionar con arcillas y otros materiales sintéticos a base de silicoaluminatos. Gran número de hidrocarburos fueron probados y los productos de reacción pacientemente identificados. De este trabajo se llegó a la conclusión de que existía una similitud en el comportamiento de los sólidos catalíticos con el de los ácidos fuertes que intervenían como catalizadores en solución. Esto permitió a los científicos especular que los centros activos que intervenían en las reacciones de desintegración de las moléculas del petróleo eran sitios ácidos. Ya en 1947 el mecanismo de las reacciones se había esclarecido lo suficiente como para proponer que se llevaban a cabo por medio de intermediarios iónicos. Se sabe hoy en día que el primer paso en estas reacciones es la formación de un ion carbonio, el cual podemos visualizar como un catión orgánico con una carga asociada al átomo de carbono. Una de las maneras como se forman los iones carbonio es por la fisión de una molécula estable en la cual uno de los fragmentos toma dos electrones y las especies resultantes forman un par iónico:
Debemos anotar que la presencia de iones carbonio había sido reportada por los químicos orgánicos en el año de 1900. Cuando se aplicó este concepto a las observaciones que se habían hecho sobre la reactividad de los hidrocarburos, así como a los productos generados, se pudo comprobar que el mecanismo concordaba perfectamente, emergiendo de ello toda una teoría, llamada del ion carbonio, con la cual es posible predecir cuantitativamente su formación y reacción, empleando las herramientas usadas en la química teórica.
Las arcillas presentan sitios ácidos de fuerza variable, tanto de tipo Bronsted como Lewis. Es obvio que la manera de identificarlos y contarlos será haciéndolos reaccionar con bases. Si conocemos la estructura de la base empleada y su fuerza relativa, entonces podemos, con la ayuda de diversas bases, hacer una especie de mapa sobre la distribución y tipo de sitios ácidos presentes. Por otra parte se sabe actualmente que las diversas reacciones catalizadas por sólidos ácidos tienen en muchos casos requerimientos específicos sobre la naturaleza y fuerza del ácido para llevarse a cabo. Por lo tanto, dependiendo de la forma de preparar el sólido catalítico y de activarlo es posible hacer concordar los requerimientos de acidez con los de reactividad.
LAS ARCILLAS Y EL ORIGEN DE LA VIDA
La búsqueda de una explicación precisa de cuáles fueron los constituyentes de partida que dieron origen a los seres vivos, es decir, aquellos sistemas capaces de reproducirse a expensas de su energía interna, ha constituido un enigma que preocupó ya a los primeros filósofos. Los hombres sabios de la antigua India, de Babilonia y de Egipto elaboraron varias cosmogonías para tratar de explicarlo. Entre las primeras destaca la que consideraba que la vida se había creado por generación espontánea, como resultado de la voluntad de fuerzas sobrenaturales. Los griegos hicieron progresos tratando el problema por métodos "científicos", aunque persistieron en creer en la autogénesis. Esta idea persistió en la Edad Media y parte de los tiempos modernos hasta que Louis Pasteur derrumbó el mito de la generación espontánea. Sin embargo, la pregunta original sigue vigente: si un organismo sólo puede ser creado por otro, ¿cómo se inició la vida?
La respuesta lógica es que la evolución química debió haber precedido a la biológica y lo demás que se añada es mera especulación. Esto ya había sido reconocido por Darwin, quien afirmaba que si en su tiempo se hubiera creado una sustancia proteica capaz de seguir experimentando transformaciones complejas, seguramente sería incapaz de hacerlo, ya que las condiciones óptimas para su evolución sólo existieron en la época en la cual no había vida. Experimentalmente es por tanto imposible reproducir las condiciones que existían en la época prebiótica y los resultados que obtengamos de dichas observaciones mostrarán únicamente probabilidades de que la vida se haya iniciado de tal o cual manera. Lo que pueden hacer los físicos y químicos es construir modelos y probar en qué grado éstos son consistentes con los hallazgos de los biólogos, geólogos, antropólogos, etc. Lo que sabemos del periodo que precedió a la vida, de acuerdo con las investigaciones geoquímicas, es que se inició de mil a dos mil millones de años atrás. La composición de la atmósfera original ha sido motivo de considerable discusión, aunque es generalmente aceptado que la mezcla actual, a base de nitrógeno y oxígeno, no pudo haber sido la original. Es muy difícil obtener evidencias, aun aproximadas, de cuáles fueron los componentes de partida pero se acepta que su naturaleza debió ser semejante a la de la nebulosa que formó nuestro sistema planetario. Un hecho parece claro: hace dos mil millones de años la atmósfera no contenía oxígeno, pero mil millones después era rica en ese elemento. En el intervalo de tiempo transcurrido entre ambos eventos emergió la vida. Muchos experimentos de laboratorio se han realizado con el fin de demostrar cómo es posible generar sustancias orgánicas a partir de compuestos inorgánicos, aun en ausencia de oxígeno. Tal vez uno de los más importantes fue el realizado por Stanley Miller en 1953. Estudiante recién graduado, Miller trató de recrear las condiciones prebióticas en un aparato de vidrio: puso unos litros de metano, amoniaco e hidrógeno para simular la atmósfera; añadió un poco de agua ("el océano") y adaptó un dispositivo capaz de generar descargas eléctricas. Después de calentar la mezcla durante algunos días analizó su contenido, encontrando varios compuestos orgánicos, notablemente aminoácidos. Después de Miller muchos investigadores han realizado trabajos similares, empleando otras sustancias de partida, demostrándose así la posibilidad de sintetizar moléculas orgánicas más o menos complejas. Baste decir que, sin importar el cómo se generaron estas primeras especies, demos por hecho de que aparecieron y que estructuralmente tenían la capacidad de seguir evolucionando por reacciones entre sí. A. Oparin, científico ruso, opina en su libro sobre el origen de la vida que las moléculas orgánicas prosiguieron su evolución hasta que, al alcanzar cierto tamaño, se separaron del medio acuoso aislándose por medio de una especie de membrana impermeable al agua. Muchos autores no aceptan este esquema de Oparin pues consideran difícil visualizar un sistema cerrado en el cual se lleva a cabo una química diferente a la del medio que la rodea sin que dicho sistema llegue a un estado de equilibrio, hecho que desde el punto de vista termodinámico implica que ya no hay variación en la energía libre y por ende, en el caso de una célula, ésta se hallaría muerta. Adicionalmente, el mecanismo no es aceptado por la dificultad de visualizar cómo unas moléculas orgánicas, flotando en un inmenso océano, hayan tenido oportunidad de encontrarse y reaccionar.
J. Bernal en su libro sobre los orígenes de la vida, publicado en 1967, opina que, dada la extrema dilución de las sustancias orgánicas, las reacciones químicas serían extremadamente lentas por lo que propone que inicialmente se llevó a cabo una concentración de las moléculas, seguida de reacciones de polimerización, es decir crecimiento de cadenas de sustancias orgánicas, todo esto llevado a cabo en las arcillas depositadas en la orilla de los océanos y lagos.
Las razones que se argumentan para esta hipótesis son las siguientes:
1) Las arcillas, por su capacidad de expandirse, pueden alojar en su interior moléculas orgánicas de gran talla.
2) El crecimiento de la estructura de las moléculas se lleva a cabo inicialmente por reacciones de polimerización, las cuales son catalizadas por centros ácidos como los presentes en las arcillas. Un punto adicional es que, en dichas reacciones de polimerización, uno de los productos de la reacción es el agua, la cual debe ser eliminada del medio para favorecer el curso de formación de los materiales orgánicos. Lo anterior es factible de realizarse en las arcillas, dada su capacidad de convertirse en materiales organofílicos.
Veamos algunos resultados de laboratorio que apoyan las tesis de Bernal y que fueron realizados con montmorillonita a la cual se le sustituyó parcialmente el aluminio original de la estructura, que es trivalente, por iones magnesio, divalentes, con lo cual se crean centros fuertemente negativos, los cuales son capaces de realizar su adsorción y reacción posterior de los materiales orgánicos. Los investigadores del Instituto Weizmann de Israel pusieron en contacto la arcilla con un aminoácido adenílico, que es el material de partida para la formacióu de proteínas en las células y cuya fórmula mostramos en la figura 27.
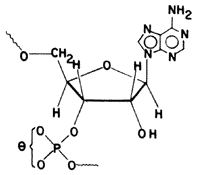
Figura 27. Estructura del aminoácido adenílico, precursor de las proteínas.
Los científicos observaron que al poner en contacto la montmorillonita con la molécula orgánica en una solución acuosa, a la temperatura ambiente, se produce primero una adsorción del material biológico y luego éste se polimeriza dando origen a cadenas polipeptídicas de diferentes pesos moleculares. Como sabemos, las cadenas polipeptídicas son las que forman las proteínas dentro de las células.
En el caso de la estructura mostrada en la figura, la reacción de polimerización se llevaría a cabo empleando la adenina, que corresponde a la molécula que contiene N (nitrógeno) en la parte superior derecha de la estructura. Esta molécula de adenina se polimeriza produciendo (adenina)n, que es el polipéptido que hemos mencionado, dando como resultado productos con valores de "n" variables. La observación interesante que hicieron los autores de este trabajo está relacionada justamente con los valores de "n". Cuando la reacción se realiza en ausencia de arcilla empleando un ácido inorgánico, se observa que los polipéptidos formados son de tamaño pequeño y su distribución muy homogénea. Por el contrario, la distribución de pesos moleculares en los productos analizados de la reacción con la arcilla muestra que la distribución es heterogénea, es decir, hay ciertos valores de "n" que no aparecen en los polipéptidos así formados. Curiosamente otros autores que han trabajado sobre el mismo tema han hecho hincapié en que los procesos vivientes se caracterizan por tener la capacidad de seleccionar cadenas de ciertos pesos moleculares específicos a pesar de que no existe impedimento para que dicha distribución sea homogénea.
De lo que hemos aprendido sobre el comportamiento de las arcillas sabemos que
el elemento inicialmente presente puede ser intercambiado por otros y así se
modifican no sólo las propiedades de adsorción sino también las catalíticas,
al variar la distribución de los centros ácidos. Esto sugeriría que la arcilla
actúa como una especie de molde prebiótico que va a determinar un código inicial
el cual, por evolución posterior, producirá selectivamente ciertos tipos de
secuencias de moléculas complejas que, ulteriormente, se convertirán en sistemas
vivientes. El apreciado lector tiene la palabra para opinar si esto pudo haber
sido o no, tomando en cuenta la forma característica de la molécula de la vida:
el ADN: una doble hélice limitada en la anchura pero ilimitada
en su longitud.
En el curso de los últimos cien años el origen del petróleo ha intrigado a geólogos, químicos, físicos y microbiólogos, sin que hoy en día exista una teoría por todos aceptada sobre su génesis. Lo anterior estriba en buena parte en que el petróleo presenta una composición química muy compleja, aunado al hecho de que en su gestación interviene un gran número de variables físicas y químicas que, dada su formación millones de años atrás, no podemos conocer con precisión. Para colmo de males el petróleo es un fluido que se origina en un lugar pero migra a otros sitios, por lo que nunca se puede afirmar categóricamente: "aquí se formó el petróleo."
La falta de "cooperación" del material en revelar todos sus secretos a los científicos no ha impedido que cada grupo que lo estudia haga especulaciones sobre su formación y, por lo tanto, existen tantas teorías como artículos hay escritos sobre el tema. Se ha sugerido que se formó por la polimerización de los gases atrapados en las rocas basálticas, reacción por cierto catalizada por los ácidos; otros afirman que la materia que le dio origen no es otra que el carbón vegetal y hasta se han avanzado teorías sobre su origen cósmico.
Una aproximación que a muchos les ha parecido útil es investigar su origen a partir de los datos que existen sobre su composición actual. Esta tarea no es nada fácil y, para ejemplificarlo, diremos que en 1927 el Instituto Norteamericano del Petróleo puso las bases de un proyecto cuyo propósito era identificar todos los componentes del petróleo, empleando los métodos analíticos disponibles. La tarea era monumental, baste decir que, 27 años después, se informaba que 141 componentes se habían aislado y purificado. Se trataba únicamente de hidrocarburos que contenían átomos de carbono e hidrógeno que iban desde uno a veinte carbones; sin embargo esta cantidad sólo representa el 44% del total de los componentes, ya que más adelante se identificaron hidrocarburos de hasta 30 átomos de carbono y combinaciones más complejas de moléculas que, además de carbono e hidrógeno, contienen nitrógeno y azufre, así como sustancias cuya estructura presenta metales como níquel o vanadio.
Gran parte de los yacimientos petrolíferos se encuentra asociada a sedimentos marinos, en los que se nota la presencia de rocas de todas las edades, de la era Cambriana hasta el Plioceno, aunque se considera que el rango de tiempo que tomó la formación del petróleo abarca unos 500 millones de años. Hoy en día existe un consenso casi general de que su origen es orgánico, derivado de materiales que fueron sintetizados por vegetales superiores en los continentes o por algas y otros organismos planctónidos en mares y océanos. Lo anterior es evidente si vemos la similitud estructural entre la molécula aislada de un vegetal y la obtenida de un crudo de petróleo tal y como se muestra en la figura 28.
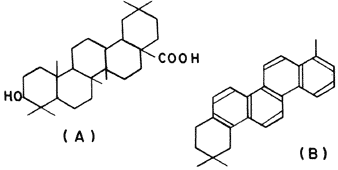
Figura 28. Estructura de una molécula aislada de un vegetal y otra de petróleo.
Si la materia orgánica que dio origen al petróleo estaba constituida por componentes provenientes de vegetales y animales, ¿cómo se transformó esa masa en una mezcla fluida con la composición que conocemos actualmente? Se cree que la materia orgánica se acumuló en vasos lacustres o marinos formando el querógeno, término que describe a una compleja molécula heterogénea, fuente del petróleo. Contrario a lo que pudiera pensarse, la transformación no se llevó a cabo a temperaturas muy elevadas. Inicialmente hubo una degradación, llevada a cabo por microorganismos, y luego la masa parcialmente degradada fue, con el curso de los años, hundiéndose en la corteza terrestre para ahí continuar su evolución a temperaturas entre 200 y 250 grados centígrados, puesto que en el análisis de crudos se ha encontrado sustancias que se descomponen a temperaturas superiores a las marcadas.
Si la masa no penetró muy profundamente en la corteza, entonces se formaron los llamados esquistos bituminosos, constituidos por rocas que contenían materia orgánica que no evolucionó, esquistos que se convirtieron en los hidrocarburos típicos del petróleo. Esto se debió a que la temperatura no fue muy elevada (se calcula que la temperatura aumenta a razón de lºC por cada 42 metros de profundidad) y sólo hubo degradación microbiana. Cuando la masa alcanzó los 200ºC ya no pudo ser degradada por seres vivos, por lo que la conversión debió proseguir a través de reacciones químicas. Paralelamente se generaron otros procesos que dieron origen a los llamados aceites pesados y arenas asfálticas; materiales geológicamente más viejos que el petróleo ya que se trata de yacimientos que se depositaron en lugares donde existían rocas porosas y que sufrieron degradación química y bioquímica adicional por la invasión de agua, proveniente de la superficie, rica en oxígeno disuelto, lo que permitió a las bacterias aeróbicas degradar selectivamente los hidrocarburos ya formados.
Terminado este esbozo de la génesis del petróleo, pasemos también a la especulación. Una pregunta que muchos investigadores se han hecho es cómo se formó la gasolina presente en el petróleo, o en forma más general, cómo se formaron los alcanos lineales. Entre paréntesis, debemos decir que la gasolina está compuesta, en parte, por alcanos, moléculas formadas por carbono e hidrógeno cuya fórmula general es Cn H2n+2 en donde n vale de uno a treinta o más. De estos hidrocarburos existen muchas variedades, los hay formando cadenas lineales o ramificadas, anillos, etcétera.
En la figura 29 se muestran algunos ejemplos de los principales tipos de hidrocarburos aislados del petróleo y se indica el porcentaje relativo de cada variedad; la letra R de las fórmulas representa grupos (C-H)n.
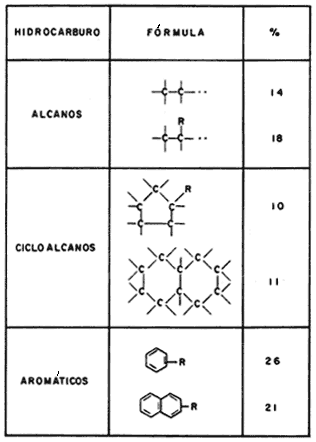
Figura 29. Algunos tipos de hidrocarburos presentes en el petróleo y su porcentaje de abundancia.
Hemos dicho con anterioridad que del estudio de los componentes del petróleo se puede obtener pistas de su origen: pues bien, estudiando la composición de los yacimientos "jóvenes" y los geológicamente más viejos y comparando los resultados con los de los crudos que se extraen actualmente, los geoquímicos orgánicos han llegado a la conclusión de que los precursores de los alcanos fueron los ácidos grasos. Se piensa además que esa transformación se llevó a cabo en presencia de arcillas.
En primer lugar, revisemos en la figura 30 las fórmulas de algunos ácidos grasos que se han aislado en el petróleo. Recuérdese que si el último H de la fórmula química de los ácidos grasos es sustituido por un ion sodio, lo que se obtiene es el conocido jabón de tocador.
|
|
|||
|
Nombre
|
Fórmula
|
||
|
|
|||
|
n-heptoico
|
CH3 (CH2)5 COOH | ||
| 2-metilcaproico | CH3 (CH2)3 CHCOOH | ||
| ½ | |||
| CH3 | |||
| 3-metilcaproico | CH3 (CH2)2 CHCH2 COOH | ||
| ½ | |||
| CH3 | |||
| 4-metilcaproico | CH3 CH2 CH (CH2)2 COOH | ||
| ½ | |||
| CH3 | |||
| 5-metilcaproico | CH3 CH (CH2)3 COOH | ||
| ½ | |||
| CH3 | |||
| 3-etilvalérico | CH3 CH2 CH CH2 COOH | ||
| ½ | |||
| C2H5 | |||
| caprílico | CH3 (CH2)6 COOH | ||
| pelargónico | CH3 (CH2)7 COOH | ||
|
|
|||
Figura 30. Variedades de ácidos grasos aislados del petróleo.
Cuando se realiza un análisis más detallado de los ácidos grasos y los alcanos presentes en los yacimientos "jóvenes" o "viejos", se obtienen los datos que mostramos en la figura 31. Marcamos en ella con (+) un valor numérico pequeño y con (+++) un valor grande.

Figura 31. Abundancia de ácidos grasos y alcanos en yacimientos: ( + ) poca, (++) mucha.
Los resultados nos enseñan que hay una tendencia, que no parece casual, en la desaparición de los alcanos lineales impares y la de los ácidos grasos pares; esto a medida que el tiempo transcurre y aumenta la profundidad a la que se encuentra el yacimiento. Es de hacerse notar que en el petróleo crudo que se extrae los alcanos y los ácidos grasos presentan distribución homogénea en pares e impares.
La explicación, según varios investigadores, consiste en pensar que el ácido graso inicialmente presente en la materia orgánica (predominantemente pares en el número de átomos de carbono) sufre una descarboxilación, es decir la pérdida del grupo -COOH, lo que da origen a un alcano:
R representa una larga cadena de grupos CH3.
El alcano formado se fragmenta a su vez (reacción de desintegración) en alcanos más pequeños
Se piensa que, en primer término, las arcillas de tipo montmorillonita concentraron en su interior los ácidos grasos por un fenómeno de adsorción, expulsando el agua estructural (es decir que se volvieron parcialmente organofílicas) y con esto se generaron centros ácidos activos que catalizaron las reacciones de descarboxilación y desintegración.
El modelo que hemos presentado se ha simulado en el laboratorio. En 1971 se publicó un informe sobre la reacción de descarboxilación del ácido graso de cadena lineal que contiene 22 átomos de carbono. Se escogió este ácido porque es típico de los yacimientos "jóvenes". El material se puso en contacto con una montmorillonita parcialmente hidratada, que contenía calcio en su estructura. Los resultados de la reacción mostraron que se producían predominantemente alcanos y, en particular, el que tenía un átomo de carbono menos que el ácido graso original.
El modelo geoquímico para el proceso que hemos descrito se puede observar en la figura 32, donde se grafica la profundidad de la corteza terrestre y la temperatura correspondiente en función de los dos tipos de reacciones que se llevan a cabo. Se postula que, al aumentar la temperatura, la arcilla genera centros ácidos del tipo Lewis, donadores de iones hidrógeno, que catalizarían dichas reacciones.
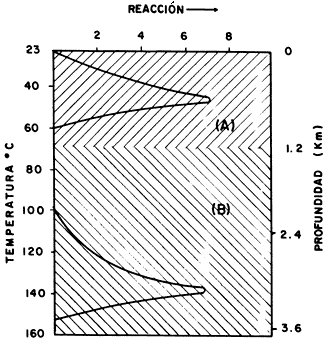
Figura 32. Modelo geoquímico para la formación de alcanos: en una descarboxilación y en una desintegración.
LAS ARCILLAS Y EL FUTURO ENERGÉTICO
En los últimos cien años la humanidad ha consumido, básicamente para proveerse de energía, unos 900 000 millones de barriles de petróleo, cantidad que equivale a quemar madera y aceites animales durante 1 300 años. Muchos esfuerzos se han realizado últimamente para racionalizar el uso del petróleo como energético, buscando por ejemplo fuentes alternas al mismo. La realidad es que para el año 2000 el 55% de toda la demanda mundial de energía será aportada aún por el petróleo. ¿Cuánto petróleo existe y para cuántos años más alcanza?, es una pregunta formulada de manera continua. Los estrategas de la planeación económica de todo el mundo hacen sus predicciones en función de las reservas de petróleo, clasificadas en dos tipos: las probadas y las potenciales.
Las reservas probadas hoy en día se estima que equivalen a 900 000 millones de barriles de petróleo y una cantidad equivalente de gas. Se trata aquí del petróleo depositado en yacimientos marinos y terrestres, explotable con las tecnologías disponibles hoy en día. Las reservas potenciales incluyen aquellas que se presume deberán descubrirse y cuya estimación es muy variable. Si se considera que el ritmo de consumo anual sea en el futuro el mismo que hoy en día, con las reservas probadas tendremos petróleo para los próximos 45 años; si sumamos las potenciales, el número de años se extiende a 170. Los economistas quedan satisfechos con sus cálculos, pero quien trabaja en la industria petrolera es más pesimista, ya que se enfrenta a futuro con dificultades técnicas para satisfacer la demanda. ¿Por qué? La respuesta la daremos con algunos datos adicionales en los que mostramos, de acuerdo con los especialistas, el tipo y porcentaje de los petróleos que se utilizan en las referidas predicciones, véase la figura 33.
|
|
||
|
Reservas (%)
|
||
|
Fuente
|
probadas
|
potenciales
|
|
|
||
|
Petróleo convencional
|
53.1
|
25.8
|
|
Petróleo pesado y arenas asfálticas
|
31.3
|
27.8
|
|
Esquisitos bituminosos
|
15.6
|
46.4
|
|
|
||
Figura 33. Origen de las reservas mundiales de hidrocarburos.
Se llama petróleo convencional al que es fácilmente transformado en productos refinados de alto rendimiento mediante los procesos y catalizadores actuales. Desafortunadamente, este tipo de petróleo ha disminuido en forma constante desde hace mucho tiempo, por lo que hay que refinar crudos más "pesados" (el crudo Maya es un ejemplo), o sea un material que, con la tecnología actual, produce mucho menos rendimiento en refinados debido a su composición, ya que posee moléculas de alto peso molecular, además de que contiene un porcentaje de azufre, níquel y vanadio superior a los crudos ligeros.
El procedimiento más empleado por la industria de la refinación para aumentar la producción de hidrocarburos gaseosos, útiles en los procesos petroquímicos de las gasolinas, diesel y lubricantes a partir de ciertas fracciones del petróleo, es sin duda el proceso de desintegración catalítica. La fracción se pone en contacto con un catalizador ácido a temperatura elevada y éste promueve la fragmentación de las moléculas, reduciéndolas a tamaños menores. Las arcillas naturales fueron usadas intensamente como catalizadores con este propósito hace unos 45 años. Básicamente se emplearon montmorillonitas tratadas con ácidos (proceso Houdry).
Si se controlaba con cuidado la intensidad del tratamiento, se obtenía un producto con buena actividad catalítica; un exceso hacía que se perdiera totalmente su función. En el tratamiento, se eliminaban principalmente iones aluminio de las posiciones octaédricas de la montmorillonita, provocando en la estructura una carga negativa que era neutralizada por el ion hidrógeno. Este hidrógeno, se cree, era la fuente de la actividad desintegradora del catalizador. Una desventaja tenían las arcillas y era la presencia en su estructura del ion hierro, el cual provocaba una excesiva cantidad de carbón proveniente de la degradación total de los hidrocarburos; además, las arcillas no eran térmicamente muy estables cuando se hacía necesario elevar la temperatura para restituir la actividad catalítica.
Las arcillas fueron paulatinamente desplazadas por otros materiales. En particular, en los años sesenta hicieron su aparición las zeolitas, que ofrecían mayor conversión, altos rendimientos en gasolina y elevada estabilidad frente a la temperatura. La estructura zeolítica, formada por bloques de SiO4 y A1O4 se une entre sí mediante puentes de oxígeno, formando estructuras que tienen poros con dimensiones moleculares y que, dependiendo del tipo de zeolita en cuestión, poseen aberturas de poros que van desde los 2 Å hasta los 8 Å. De esta manera la zeolita tiene la capacidad de permitir o no la entrada de reactivos en el interior de su estructura y llevar a cabo las reacciones ácidas con suma eficiencia.
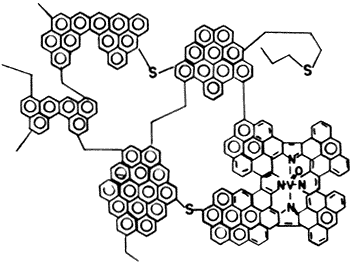
Figura 34. Estructura de una molécula aislada del petróleo pesado, que contiene vanadio.
La necesidad de refinar crudos pesados trae consigo la dificultad de hacerlo con los materiales zeolíticos de que disponemos hoy en día: la abertura de los poros en las zeolitas es mucho menor que el tamaño de muchas de las moléculas presentes en los crudos pesados. Además, las altas concentraciones de metales como el níquel y el vanadio las inactivan o, peor aún, las destruyen. En la figura 34 mostramos una versión simplificada de la estructura de una de las moléculas que contienen vanadio y que se hallan en los crudos pesados, a la que se determinó mediante estudios de rayos X. Para resolver el problema de la refinación de los crudos actuales, se han mencionado varios tipo de estrategias, las más importantes son las siguientes:
1) Emplear zeolitas con abertura de poro mayor a las que hoy empleamos. A nivel mundial se intenta sintetizar afanosamente nuevas estructuras que posean esta característica sin que pierda la activad catalítica.
2) Fragmentar el crudo con otro tipo de catalizadores que produzca moléculas capaces de penetrar las cavidades zeolíticas.
3) Buscar otro tipo de materiales porosos con alta actividad y selectividad apropiada. En este caso, las arcillas expandidas resultan ser los candidatos idóneos como también lo son para la estrategia precedente.
Veamos qué sucede cuando una fracción de crudo pesado se hace pasar por un reactor donde hay un catalizador, a la temperatura de 560° C. En el experimento se utilizarán dos tipos de sólidos: el primero estará constituido por un catalizador de desintegración a base de zeolita, al que llamaremos "CZ". En el segundo experimento emplearemos un catalizador mixto, compuesto de 20% de zeolita y 80% de arcilla del tipo montmorillonita expandida con un oligómero de aluminio, y que llamaremos "CAZ". La corriente que emerge del reactor es analizada en sus componentes, con lo cual se puede determinar qué porcentaje de la carga se convierte en productos (% Conv.) y de lo convertido, cuánto es gasolina y aceite ligero, lo que denominamos selectividad, Sgasolina y Saceite ligero respectivamente.
Observemos los resultados de las dos experiencias en la figura 35.
|
|
||
|
Catalizador
|
||
|
Parámetro
|
CZ
|
CAZ
|
|
|
||
|
Conversión del crudo
|
57.2
|
72.1
|
|
Selectividad gasolina (%)
|
27.6
|
30.4
|
|
Selectividad aceite ligero (%)
|
15.1
|
12.7
|
|
|
||
Figura 35. Conversión catalítica de un crudo mediante catalizadores a base de zeolitas (CZ) y de zeolita + arcilla (CAZ).
Como se puede apreciar, la estrategia parece dar resultado ya que la adición de la arcilla al catalizador de zeolita hace que aumente la conversión del reactivo así como la cantidad de gasolina producida, por lo que se concluye que las grandes moléculas son fragmentadas en la arcilla y luego pasan a la zeolita en donde se continúan las reacciones de desintegración.
Hemos mencionado antes que muchos crudos pesados contienen cantidades importantes de compuestos orgánicos de níquel y vanadio. Las arcillas expandidas ofrecen la posibilidad de atrapar estas moléculas por medios catalíticos, impidiendo así que dañen los catalizadores de zeolita. Para eliminarlos se requiere que el sólido contenga dos funciones: una ácida, que provoque la ruptura de los enlances, y otra capaz de introducir hidrógeno en los fragmentos resultantes, ya que si no se hidrogenan totalmente tienden a formar residuos carbonosos que inhabilitan la actividad catalítica. La función hidrogenante en el catalizador la aporta un metal depositado ex profeso.
En una serie de experimentos se expandió una arcilla de tipo montmorillonita con un oligómero orgánico a base de hierro y una muestra adicional con uno de aluminio. Los materiales fueron evaluados por separado, haciéndolos pasar en un reactor, durante 72 horas, una carga consistente en un crudo con cantidades conocidas de níquel y vanadio. Los resultados mostraron que la arcilla expandida con aluminio tuvo muy poca capacidad de eliminación, como era de esperarse, por no tener la función hidrogenante. En la figura 36 se resumen los resultados obtenidos con la arcilla que contiene hierro, en la que se grafica el porcentaje de metal eliminado de la carga en función de la temperatura de reacción.
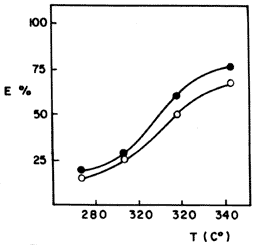
Figura 36. Eliminación porcentual de vanadio (![]() )
y níquel (O) mediante un tamiz de arcilla en función de la temperatura de reacción.
)
y níquel (O) mediante un tamiz de arcilla en función de la temperatura de reacción.
Cuando estos datos se confrontan con los que se obtienen con los catalizadores actualmente empleados en las refinerías, que son de naturaleza química diferente, se comprueba que si bien el porcentaje de eliminación es semejante, la arcilla realiza la tarea a temperatura inferior, lo que representa un ahorro en energía.
Hemos resaltado a lo largo de este capítulo que una de las ventajas destacables de las arcillas modificadas frente a otros sólidos porosos la constituye su capacidad de adsorber y hacer reaccionar en el interior de su estructura moléculas de gran tamaño.
Como último ejemplo del potencial que ofrecen estos nuevos materiales, presentaremos algunos resultados obtenidos por J. Shabtai y sus colaboradores que fueron tratados en el 7° Congreso Internacional de Catálisis efectuado en Japón. Los autores del trabajo hicieron un estudio comparativo de la reactividad de tres diferentes moléculas orgánicas frente a una zeolita Y, la que se emplea en los catalizadores en la desintegración de los gasóleos y una montmorillonita expandida. Se sabe que la abertura máxima de los poros zeolíticos tiene un diámetro aproximado de 8 Å y las tres moléculas escogidas diámetros de 6.2, 7.5 y 9 Å. Estos diámetros de las moléculas, hemos de aclarar, que son valores absolutos y se calculan a partir de estimaciones teóricas. Sin embargo tal es quizá el método más práctico que se tiene para medir la abertura de los poros. El experimento consistió en evaluar la velocidad de transformación por minuto del reactivo, referida a una masa idéntica de sólido en cada caso. La reacción en que se emplearon las moléculas sonda fue realizada a 350° C y con el fin de exponer los resultados no es necesario dar los valores absolutos de tales velocidades, así, en el caso de la zeolita le daremos el valor relativo de 1 (Vr); en el de la arcilla, el número de veces que la velocidad de transformación fue más rápida que 1.
En la figura 37 se representan los resultados. Se dibujó la estructura de las moléculas-reactivo empleadas y, entre paréntesis, va anotado el diámetro que se les calculó en Angstroms.
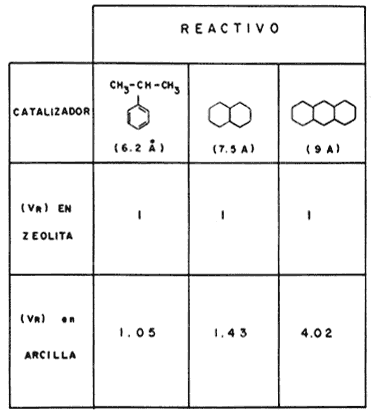
Figura 37. Comparación entre las velocidades de reacción (V) en la conversión de tres reactivos con distinto diámetro cinético.
Es evidente que, al aumentar el tamaño de la molécula, la zeolita tiene más dificultad para convertirla por lo que la velocidad de reacción de la arcilla es cada vez mayor ya que en ella no existe el problema de que el reactivo no penetre al interior de la estructura.
Debemos mencionar que el trabajo fue presentado en 1980 y que, en la sección de comentarios del Congreso, el representante de una industria petrolera dijo: "Quiero felicitarlos por este trabajo tan innovador, realizado con tanta imaginación. Tengo la impresión de que esto representa el punto de arranque de nuevos desarrollos que se iniciaron hace años con los catalizadores zeoliticos." El comentarista no estaba equivocado pues podemos imaginar el potencial que tienen los valores de la figura si tomamos en consideración que, en esa época, un tercio de los cinco millones de barriles de petróleo se enviaba diariamente a procesar mediante la reacción de desintegración catalítica.