VI.LA BIOLOGÍA ESTRUCTURAL Y EL ADN RECOMBINANTE: EN LA FORMA ESTÁ LA CLAVE
AL CONTEMPLAR el complejo funcionamiento celular en el cual los genes conciertan la interacción de miríadas de moléculas, donde se transforman sustancias de manera intrincada, vemos aparecer forma y función. ¿Dónde se localiza el secreto mas íntimo de este proceso? ¿Cómo se encuentran las moléculas unas con otras? ¿A qué se debe que una enzima sea capaz de localizar y transformar la sustancia sobre la cual actúa? Las respuestas a estos interrogantes se derivan, en su forma más contundente, de la forma espacial de las moléculas. La disciplina que desarrolla y utiliza técnicas experimentales para averiguar la estructura tridimensional de las moléculas biológicas, la biología estructural, constituye una de las vertientes más importantes de la actividad científica moderna. Ya se mencionó, en el capítulo II, que fue precisamente el conocimiento estructural el que inició la revolución de la genética molecular.
En este capítulo revisaré algunos de los conceptos y desarrollos más interesantes del campo. El tema es, para mí, particularmente atractivo: en mi línea de investigación utilizamos los datos de la estructura de las proteínas y el ADN recombinante para generar nuevas versiones, con atributos mejorados, de las proteínas naturales. Es mi sincero deseo el poder transmitir al lector el sentido estético y el enorme potencial que se originan del conocimiento de las estructuras moleculares biológicas.
Si revisamos nuestras primeras experiencias con el microscopio, durante nuestras clases de biología, quizá recordemos que para lograr ver cosas más y más pequeñas, había que tomarse más y más trabajos. Para amplificar cien veces la muestra sólo necesitábamos alumbrar y enfocar bien; pero si se trataba de amplificarla mil veces, entonces usabamos aceite de inmersión para acercarnos más a la muestra; las cosas se ponían difíciles. Quizá recordemos también al maestro de biología diciéndonos que para ver cosas aun más pequeñas, la luz ya no funciona. En efecto, la longitud de onda de la luz visible tiene una magnitud tal que no permite continuar los procedimientos ópticos de manera indefinida. Recurrimos así al microscopio electrónico. De esta forma ya podemos ver estructuras dentro de la célula; ¡lo que la luz no alcanzaba, los electrones si revelan! Desafortunadamente, el mismo principio que limita la utilización de la luz, también la limita con los electrones. Los átomos y las moléculas no se pueden ver directamente.
Para deducir la estructura espacial de la materia, la manera como se asocian unos átomos con otros en tres dimensiones, requiere aplicar otros desarrollos de la física: la cristalografía de rayos X y la resonancia magnética nuclear (RMN). A partir del conocimiento íntimo de la naturaleza de los átomos y la radiación electromagnética, en los últimos 50 años se han desarrollado técnicas de increíble complejidad y elegancia, que permiten deducir, a partir de numerosas observaciones experimentales, la posición precisa de los miles de átomos que constituyen una molécula de proteína, o hasta de las muchas moléculas que constituyen un virus completo.
Aunque está fuera de los propósitos de este libro una descripción técnica de estos procedimientos, destacaré algunos de sus rasgos más importantes.
Los rayos X interaccionan con las sustancias desviándose ligeramente. La magnitud de esta desviación depende de la densidad electrónica que encuentren a su paso. Por esto los materiales que contienen átomos de metales (por ejemplo, el plomo o el calcio de los huesos), muestran zonas oscuras en una radiografía, dado que los metales son muy densos en electrones. Los rayos X también se dispersan o difractan cuando atraviesan materia orgánica, pero más ligeramente. En principio, como el patrón de difracción resultante del paso de los rayos X depende de la estructura de la materia que atraviesa, podríamos inferir ésta de aquél. El problema es que no podemos observar el patrón de difracción de una sola molécula: la señal seria demasiado débil. Para solucionar este problema se recurre a la observación de los patrones de difracción generados por cristales. Cuando hablamos de cristal, de manera rigurosa nos referimos a un fenómeno natural realmente bello. Las moléculas que pasan suavemente de estar disueltas al estado sólido, lo pueden hacer acomodándose ordenadamente una al lado de la otra, en una látice o arreglo tridimensional. La manifestación macroscópica de este fenómeno es la apariencia pulida y transparente que tienen los cristales, y sus formas geométricas características. Pero internamente, existe un orden perfecto. Todas las moléculas se encuentran orientadas de la misma manera. Así pues, al manipular un cristal de dimensiones macroscópicas (por ejemplo un milímetro), estamos manipulando de manera concertada todas las moléculas que lo constituyen, obteniendo la misma orientación de cada una de ellas respecto al observador. ¡O a un haz de rayos X! Es así como, disponiendo de cristales, se pueden obtener patrones de difracción de rayos X observables, porque provienen de muchas moléculas, y coherentes, porque las moléculas están todas igualmente orientadas respecto al rayo. De estos patrones de difracción puede inferirse la estructura individual de las moléculas que conforman los cristales.
Por desgracia, esto es más fácil decirlo que hacerlo. Para empezar hay que ser capaz de obtener los cristales. Aunque se ha logrado hacer que cristalicen gran número de moléculas interesantes, en el caso de las proteínas, de crucial importancia, no es nada fácil. La labor del cristalógrafo implica además muchos pasos posteriores a la cristalización. Hay que colectar datos, preparar derivados de los cristales originales, reducir los datos mediante complejas ecuaciones, preparar un modelo molecular que se ajuste a estos datos y refinar el modelo en muchos intentos sucesivos.
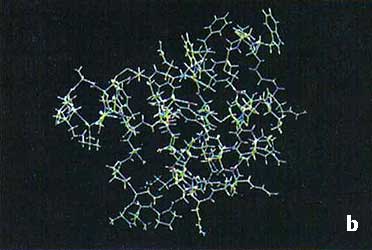
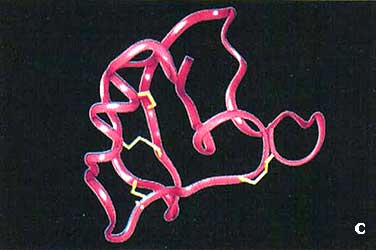
Algunas de las imágenes más bellas que ilustran los libros de bioquímica y biología molecular provienen del arduo trabajo de los cristalógrafos. Y su utilidad también es enorme: del análisis de estas imágenes tridimensionales se derivan el secreto de las interacciones moleculares, de la vida misma.
La primera estructura tridimensional de una proteína fue obtenida mediante el trabajo pionero de Max Perutz, quien requirió de 22 años para lograr su objetivo. Incluso, hasta hace poco más de una década, la resolución de una estructura proteica podía considerarse una empresa para buena parte de la vida. Afortunadamente, el avance de la metodología ha sido impresionante. Hoy día se conjugan varios factores que permiten un flujo continuo de nuevas estructuras. En primer lugar, como puede entenderse fácilmente, la potencia y abaratamiento de las computadoras modernas simplifica y acelera significativamente el trabajo de cristalografía.
Adicionalmente, la posibilidad de clonar y sobreexpresar genes ha abierto la puerta para trabajar con proteínas que antes resultaban inaccesibles. Los cristalógrafos se han asociado también a los sincrotrones, construidos para investigaciones de física atómica, pero que generan como subproducto rayos X de gran intensidad y versatilidad. El ciclo en el que los avances científicos y tecnológicos se potencian unos a otros se observa con claridad: son precisamente los últimos 10 o 15 años en los que ha registrado un impresionante avance en la biología estructural. Justo en este periodo surgió el ADN recombinante y el crecimiento explosivo de la informática.
En los últimos años ha madurado otra técnica que permite también deducir la estructura molecular: la resonancia magnética nuclear; que aplicada a proteínas, complementa la cristalografía de rayos X.
La adaptación de la RMN al trabajo con moléculas grandes surgió de avances tecnológicos recientes. Nuevamente, se requiere colectar y procesar cantidades masivas de datos. Además, se necesitaron aparatos que trabajaron con frecuencias de ondas de los que antes no se disponía. Los nuevos aparatos requieren el uso de superconductores para generar los campos magnéticos. La gran virtud de la RMN es que puede usarse para analizar moléculas en solución, es decir, en su ambiente natural. La naturaleza de la técnica es tal, que no es importante que todas las moléculas estén orientadas de la misma manera.
Con la RMN los datos observados se traducen en una serie de distancias y ángulos entre los átomos. A partir de esta lista de ángulos y distancias el espectroscopista se da a la tarea de construir un modelo molecular que satisfaga las medidas observadas experimentalmente.
El esfuerzo de los investigadores de este campo fue distinguido con el Premio Nobel de química en 1991, otorgado al investigador suizo Richard Ernst y los límites de la técnica se han extendido grandemente. Por desgracia, este método no permite determinar la estructura de moléculas grandes, tales como muchas proteínas y agregados macromoleculares.
Las técnicas experimentales para la determinación de estructura han generado un número muy importante de resultados. Hoy disponemos de más de 5 000 diferentes estructuras de ácidos nucleicos y proteínas, que representan cerca de 500 diferentes arquitecturas (el resto está constituido por variantes de las mismas proteínas, por ejemplo, provenientes de diferentes organismos o interactuando con sus moléculas blanco, etc.). En el recuadro Vl.1 se muestran representaciones esquemáticas de algunas de la estructuras de biomoléculas determinadas hasta la fecha. Desafortunadamente, esta información está cada día más rezagada de los datos conocidos sobre secuencia (véase el capítulo IV). Esto genera una urgente necesidad de desarrollar sistemas para predecir la estructura tridimensional a partir de datos de secuencia.

LA PREDICCIÓN DE LA ESTRUCTURA DE LAS PROTEÍNAS
Hace más de dos décadas, el investigador danés Christian Anfinsen —quien obtuvo el Premio Nobel de química en 1972— estableció que la estructura tridimensional de algunas proteínas está determinada por su secuencia lineal de aminoácidos. Hoy día sabemos que éste es el caso de la mayoría de las proteínas, quizá de todas. Esta convicción hace verdaderamente irresistible el reto de desarrollar métodos que nos permitan predecir su estructura a partir de la secuencia De entrada dejaremos en claro que éste es un problema que se considera algo no resuelto. Se ha dicho que el código que relaciona la secuencia con la estructura es como el segundo código genético. Realmente es a través de la estructura tridimensional de las proteínas, no directamente de su secuencia, que los genes orquestan toda la actividad celular. Este es, pues, uno de los grandes problemas pendientes de la biología moderna.
Quizá algunos lectores se pregunten: ¿las poderosísimas computadoras que existen en la actualidad son incapaces de programarse para resolver este problema? El asunto es que este problema tiene variadas e importantes aristas. Por una parte existe un número astronómico de maneras diferentes de arreglar los átomos de una proteína en el espacio. Por la otra, las fuerzas de atracción y repulsión que determinan que una proteína se pliegue en el espacio de una manera específica son muy numerosas y con diferencias de magnitud muy sutiles.
Para algunas corrientes, la predicción de la estructura de proteínas no podrá lograrse por muchas décadas. Esta visión se refiere quizá a la solución rigurosa del problema. Para otros, algunas de las aproximaciones recientes constituyen avances que permiten prever resultados concretos y prácticos en muy corto plazo.
En los últimos 5 años se han desarrollado técnicas muy elaboradas e ingeniosas que permiten extraer y codificar mucha de la información presente en la base de datos de estructuras tridimensionales conocidas. De aquí se derivan aproximaciones empíricas, que no presuponen que se entiendan finamente las fuerzas que intervienen en el fenómeno de plegamiento. De esta manera, el enfoque más promisorio, en la actualidad, tiene como objetivo inicial decidir con cual de las estructuras tridimensionales ya conocidas es compatible la secuencia de una proteína problema. Lo interesante de la situación es que podemos predecir que existen sólo alrededor de mil o menos posibles arquitecturas proteicas naturales. Todas las proteínas existentes se plegarían en el espacio en alguna de estas arquitecturas. Este es un dato muy interesante si consideramos que en un ser humano se calcula que existen alrededor de 100 mil proteínas diferentes. La predicción mencionada parte de un análisis estadístico de la frecuencia con la que los cristalógrafos y espectroscopistas obtienen estructuras que representan nuevas arquitecturas. Resulta ser que con relativa frecuencia la estructura de una proteína recientemente resuelta se parece mucho a la de alguna que ya existía en la base de datos.
Con la constante refinación y el avance de los métodos de predicción y de las herramientas computacionales que éstos utilizan, esperamos tener una idea aproximada de la estructura espacial de muchas de las proteínas cuya secuencia aparezca como fruto de la secuenciación del genoma humano, que podría completarse dentro de unos 10 años (véase el capítulo IV).
APLICACIONES DE LA INFORMACIÓN ESTRUCTURAL
Quizá muchos lectores coincidan en que el conocimiento de las estructuras que adoptan las biomoléculas constituye un logro espectacular de la ciencia moderna. En cada época, los biólogos estructuralistas han utilizado los últimos adelantos tecnológicos para avanzar en su trabajo, y así han ido desentrañando detalladamente la estructura de moléculas complejísimas. Cabe preguntarse sin embargo: ¿de dónde surge la motivación para dedicar décadas enteras a dilucidar determinada estructura molecular? Ciertamente, el placer estético de observar la estructura resuelta no parece ser una justificación suficiente para la inversión de tiempo y recursos necesarios. La respuesta, que con gran intuición destacaron los pioneros de la biología estructural, es que en la estructura detallada de las macromoléculas biológicas se encuentran los secretos más íntimos de los fenómenos de la vida.
Pensemos qué ocurre cuando ingerimos un medicamento y éste ejerce su efecto.
El medicamento es una sustancia química que tiene afinidad por alguna de las
macromoléculas (generalmente una proteína) de nuestro organismo. La sustancia
medicinal entra al organismo, viaja por medio del torrente sanguíneo, penetra
en las células y finalmente encuentra una molécula blanco. La unión específica
entre estas dos sustancias da como resultado el efecto medicinal. Lo verdaderamente
sorprendente es que hasta la fecha no tenemos una idea clara de los detalles
de estas interacciones. Como ya hemos señalado (véase el capítulo anterior;
"El valor de la biodiversidad en el contexto de la biotecnología moderna") casi
la totalidad de las medicinas usadas actualmente provienen de principios activos,
aislados de plantas, cuya utilidad fue encontrada empíricamente, quizá preservada
por la tradición de muchos siglos. ¿Qué podríamos esperar, sin embargo, si conocemos
la estructura detallada de las moléculas relevantes para determinada enfermedad?
Lo promisorio del conocimiento estructural en este campo es que se puede pensar
en diseñar los medicamentos específicamente para unirse a sus moléculas blanco.
Habrá quien declare que hasta el momento esto se ha podido lograr sólo en casos
excepcionales. Aunque cierto, otros tenemos la convicción de que tarde o temprano
el diseño racional de medicamentos será la norma y no la excepción. De hecho,
todas la compañías farmacéuticas del mundo invierten cuantiosas sumas de dinero
en investigación sobre biología estructural y en diseño de moléculas por computadora.
Aun cuando quizá estos desarrollos no alcancen a beneficiar a muchos de los
enfermos de hoy día, el incontenible avance de las técnicas empezará a transformar
el ejercicio de la farmacología en un futuro tal vez más cercano de lo que imaginamos.
Un notable ejemplo entre varios que se desarrollan en la actualidad es la investigación
que se realiza para contrarrestar el devastador efecto del virus del SIDA,
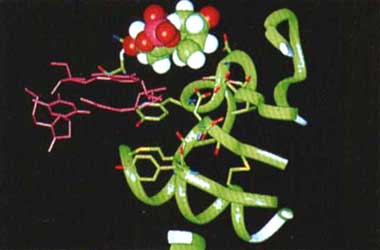
como se muestra en el recuadro VI. 2.
BIOCATÁLISIS E INGENIERÍA DE PROTEÍNAS
Otro importante desarrollo relacionado con la estructura de macromoléculas es la biocatálisis. Consideremos la increíble variedad de materiales y procesos que se observan en el mundo viviente. Todavía no ha sido capaz el hombre de superar las propiedades de fibras como la lana o el algodón o de fabricar zapatos con materiales que mejoren las cualidades del cuero. Seguimos utilizando perros para detectar cantidades minúsculas de drogas, porque no existe un aparato que tenga tal sensibilidad y capacidad de discriminación. Estos logros son producto de 3 500 millones de años de evolución, pero el conocimiento preciso de las propiedades de los sistemas vivos nos permitirá, cada vez más, inspirarnos con las formas como la naturaleza ha resuelto los retos de la supervivencia, y aprovechar la infinidad de materiales que ella ofrece para diseñar productos o procesos específicamente útiles para las actividades y el bienestar humano.
En particular, la capacidad catalítica de los sistemas vivos supera con amplísimo margen la que ha logrado la industria química. Actualmente los productos químicos se producen mediante reacciones que utilizan toda clase de diferentes condiciones de temperatura, presión, uso de disolventes, etc. Se purifican con grandes aparatos de destilación, filtración, etc. En contraste, los seres vivos realizan también toda clase de reacciones químicas y producen toda clase de sustancias y materiales, pero siempre a temperaturas moderadas y en solución acuosa. Nuevamente, podemos pensar en utilizar estrategias y materiales similares a los biológicos para realizar las transformaciones químicas que nos interesan. Aquí el conocimiento estructural es extremadamente importante. Consideremos que normalmente una enzima está adaptada para trabajar dentro de una célula, sujeta a los requerimientos de regulación y recambio que ésta requiere. Además, efectuará una reacción química específica, parte del metabolismo celular. En las aplicaciones tecnológicas normalmente requeriremos actividades catalíticas más o menos distintas a las naturales. Necesitamos también enzimas estables y fáciles de obtener en grandes cantidades. Como fruto del esfuerzo de varias décadas se ha logrado "domesticar" cierto numero de enzimas para el servicio del ser humano. Por ejemplo, los detergentes biológicos contienen proteasas (enzimas que catalizan la degradación de proteínas) y lipasas (que degradan grasas), que hace más eficiente el lavado. En la industria del refresco se utilizan los jarabes con alto contenido de fructosa que se producen a partir de almidón, mediante la degradación y conversión enzimática de dos productos edulcorantes básicos: glucosa y fructosa. Inclusive se utilizan procesos de biocatálisis para la producción de compuestos de petroquímica secundaria, como el antiespumante acrilamida. Estos logros han sido posibles aun sin la capacidad de cambiar de manera importante la actividad enzimática básica encontrada en la naturaleza.
Hoy día, sin embargo, basándonos en las estructuras tridimensionales de las
enzimas, podemos aplicar las técnicas del ADN recombinante para
modificar significativamente las propiedades de las enzimas, para crear biocatalizadores
hechos a la medida. Ha surgido una nueva disciplina: la ingeniería de proteínas.
Los procedimientos implicados son conceptualmente simples: se aísla el gene
que codifica para la enzima de interés; se introducen cambios en su secuencia
mediante el uso de ADN sintético (véase en el capítulo II, "El
ADN sintético"); se sobreproduce la proteína en algún sistema de
expresión. Claramente, aunque las técnicas de ADN recombinante
permiten fabricar genes con la secuencia que se nos antoje, se requiere un conocimiento
de la estructura tridimensional de la proteína codificada para poder diseñar
racionalmente los cambios. Precisamente con base en estos conocimientos ha sido
posible alterar las propiedades de muchas proteínas para hacerlas más útiles.
Un caso digno de mencionar es la alteración de las propiedades farmacológicas
de la insulina humana. Como ya mencionamos (capítulo V), la producción de insulina
humana por bacterias fue uno de los primeros logros de la ingeniería genética.
La utilidad de esta hormona es limitada, sin embargo, porque al no producirse
de manera continua por las células del páncreas, dentro del propio organismo,
su disolución y concentración en la sangre no sigue los patrones más adecuados.
Se requiere disminuir la tendencia que tiene la molécula natural a agregarse
entre sí. Con base en el conocimiento de la estructura tridimensional de la
insulina (determinada hace varias décadas) fue posible diseñar cambios que se
localizan precisamente en la interfase entre las dos moléculas que se agregan,
y que, al alterar su carga eléctrica, las hace repelerse, haciéndolas más solubles.
Otros muchos ejemplos atestiguan la creciente capacidad de cambiar los atributos
de las proteínas mediante el diseño racional.
EVOLUCIÓN DIRIGIDA O DISEÑO "IRRACIONAL" DE MOLÉCULAS
La ingeniería de proteínas, sin embargo, no está condicionada totalmente al
avance de nuestros conocimientos. En este momento se desarrollan también enfoques
muy interesantes para llegar más lejos que lo que nuestro conocimiento permitiría.
Sabemos que la evolución ha producido seres de pasmosa complejidad, por medio
de un proceso ciego de variación y selección. ¿Qué pasaría si pudiéramos simular
estos procesos evolutivos, sólo que en tiempos mucho más cortos? Una vez más,
el ADN recombinante permite realizar experimentos antes imposibles.
Utilizando ADN sintético y enzimas que lo reproducen (véase en
este capítulo, "La predicción de la estructura de las proteínas"), podemos generar
numerosísimos conjuntos de moléculas diferentes.
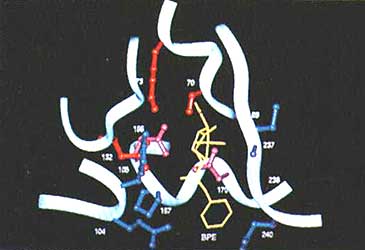
Figura VI. 1 (véase más adelante)
Al explicar los conceptos de la evolución molecular dirigida tengo la oportunidad (y perdóneme el lector que la aproveche) de explicar el trabajo que realizamos en mi laboratorio del Instituto de Biotecnología. Desde que tuve la oportunidad de viajar a Estados Unidos para especializarme en las técnicas de síntesis química de oligonucleótidos me llamó poderosamente la atención que los oligos podían utilizarse para alterar específicamente genes particulares. Más adelante decidí especializarme en utilizar estas técnicas para realizar mutagénesis a saturación y mutagénesis combinatoria. Estos conceptos se refieren al proceso de generar variabilidad dentro de un gene. Pero veamos con más detenimiento de qué se trata.
En un procedimiento tradicional de mutagénesis dirigida por oligonucleótidos
(véase el recuadro II.4. El ADN sintético y sus aplicaciones) se
introduce un cambio predeterminado, codificado en la secuencia del oligo, al
gene objetivo. ¿Qué pasaría, sin embargo, si un esquema similar se utilizara
pero con una multitud de oligonucleótidos, cada uno con una secuencia un poco
diferente? La respuesta es que obtendríamos un conjunto de moléculas variantes
en la zona afectada por el oligo. Es importante darse cuenta de que cuando hablamos
de la obtención de "un oligo", o de la donación de "un" segmento de ADN
en "un" vector de donación, en realidad hablamos de manipular trillones
de moléculas simultáneamente, todas ellas iguales. Pero podemos hacer que estas
moléculas no sean exactamente iguales. Pensemos que al sintetizar un oligo,
en una determinada posición no adicionamos sólo la base correspondiente a la
secuencia natural, sino que ponemos una mezcla de las cuatro bases. Supongamos
que el oligo llevaba ya adicionadas unas 15 bases, con una secuencia determinada.
A partir de la base 16, las cadenas de oligo que van creciendo en la resma de
síntesis ya no son todas iguales. Ahora tenemos cuatro tipos: todos los oligos
son iguales en sus primeras 15 bases, pero hay unos que contienen A, otros G,
otros C y otros T, en la posición 16. Tenemos cuatro tipos de oligos. Si repetimos
este procedimiento, los oligos resultantes serán de 16 clases distintas, cuatro
que ya existían, multiplicadas por cuatro nuevas posibilidades. La continuación
de este proceso genera toda una serie de combinaciones de secuencias en la región
donde se adicionan las cuatro bases. Después, se puede regresar al régimen anterior;
y adicionar bases únicas en secuencia definida. Obtendríamos así, adheridos
a la resma de síntesis, desde unos cuantos hasta trillones o más de oligonucleótidos,
todos con extremos iguales, pero que en el centro son diferentes. Al incorporar
oligos de esta mezcla (llamados oligos degenerados) en un protocolo de mutagénesis
dirigida (véase nuevamente el recuadro II.4), obtendríamos desde unos cuantos
hasta trillones de diferentes genes variantes, que luego podemos introducir
a las células para su expresión.
Existen distintas maneras de inducir variabilidad en moléculas de ácidos nucleicos, pero todas tienen en común los mismos elementos. Una vez obtenida una variante, su replicación perpetúa el cambio inducido. La expresión del gene, dentro de una célula, produce una proteína distinta para cada variante, de acuerdo con el código genético. Resulta así que la manipulación de ácidos nucleicos ofrece un mecanismo relativamente simple (infinitamente más simple que el de la síntesis orgánica directa) para crear innumerables moléculas distintas. El siguiente paso, tal y como nos lo ha indicado la evolución natural, consiste en diseñar un método de selección. Hay que detectar e identificar cuál de los genes variantes dio origen a una proteína con una actividad interesante. Para ejemplificar cómo se puede lograr; citaré un caso ocurrido en mi laboratorio.
Estamos interesados en explorar la relación entre la estructura y la función
de una enzima llamada ß-lactamasa, que producen diversas bacterias para
defenderse de la penicilina. La ß-lactamasa nos llamó la atención en cuanto
se publicó su estructura tridimensional (obtenida por cristalografía de rayos
X). Lo atractivo de esta enzima es que su actividad consiste en degradar la
penicilina y, al hacerlo permite que una bacteria que expresa ß-lactamasa
pueda vivir en presencia de este antibiótico (note el lector que es precisamente
el gene que codifica para ß-lactamasa el que se utiliza abundantemente en
los vectores de donación bacteriana, ya que permite detectar las células que
han adquirido el plásmido; véase el recuadro II.2. Los procedimientos básicos
del ADN recombinante). Esto quiere decir que tenemos disponible
de inmediato un método de selección.
¿Qué preguntas podemos responder aplicando los conceptos de variación y selección mencionados antes sobre la ß-lactamasa? Hemos enfocado nuestros experimentos en el problema de la especificidad de esta enzima. La ß-lactamasa con la que trabajamos es capaz de reconocer y destruir penicilinas, pero no cefalosporinas, que son compuestos muy similares. ¿En qué parte y de qué manera determina la secuencia de aminoácidos de la ß-lactamasa el que sea específica para penicilinas? Para responder esta pregunta utilizamos la información proporcionada por la estructura cristalográfica. Observamos que en el sitio activo se encuentra una serie de aminoácidos que interaccionan con la penicilina (véase la figura VI. 1). Una vez identificados estos aminoácidos potencialmente involucrados en el reconocimiento de la penicilina, diseñamos experimentos para introducir variabilidad en la posición que ocupan éstos dentro de la enzima. Mediante oligos degenerados, como los descritos anteriormente, generamos millones de diferentes genes de ß-lactamasas, que difieren entre sí en alguno o algunos de los tripletes de bases que corresponden a los aminoácidos seleccionados. A partir de una biblioteca de clonas con esta colección de genes, podemos identificar una bacteria que ahora crezca en la cefalosporina, debido a que la ß-lactamasa ya la puede reconocer.
Hasta el momento, en el laboratorio hemos encontrado un buen número de ß-lactamasas variantes que reconocen diversos antibióticos. Con los datos obtenidos hemos aprendido mucho acerca de la relación entre la estructura y la función de estas enzimas. Adicionalmente, contamos con un método experimental para evaluar para qué antibióticos, de tipo de las penicilinas y cefalosporinas, es más difícil obtener una ß-lactamasa que los destruya. Esta información puede ser de utilidad para las compañías farmacéuticas que desarrollan estos antibióticos.
La extensión natural de este tipo de investigación es la obtención de nuevas enzimas cuyas actividades y especificidades se adapten a las necesidades humanas. En particular, esquemas como el que estamos desarrollando pueden ser adaptados para la obtención de biocatalizadores, que tienen un gran potencial en la industria de la química fina (incluida la industria farmacéutica).
La explotación de los métodos de generación de variabilidad, mediante el uso de ADN recombinante, augura un futuro muy promisorio para obtener moléculas hechas a la medida. Se puede decir que en los últimos cinco años, usando estos métodos, se han explorado las propiedades de más compuestos que en toda la historia.