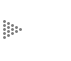III. LA FÍSICA Y LA QUÍMICA DE LA CORROSIÓN. LA CORROSIÓN METÁLICA COMO CIENCIA APLICADA
YA EN EL capítulo II habíamos mencionado que algunos metales son más activos que otros debido a la energía acumulada que poseen. En el presente capítulo haremos hincapié en la naturaleza de dichas energías, su cuantificación y los efectos que acarrean durante un proceso de corrosión.
La experiencia nos ha enseñado que todos los metales que conocemos poseen diferentes tendencias a corroerse. Es decir, unos serán más resistentes que otros a reaccionar frente a un mismo medio. Ejemplo de esto son el oro y el hierro en agua dulce. El primer metal posee una alta inmunidad frente al agua, conservando inalteradas todas sus propiedades físicas y químicas. En cambio el hierro se degrada y poco a poco se transforma en otro compuesto. El cuestionarnos el porqué de tal diferencia entre uno y otro metal, es regresar a los orígenes automáticos de los elementos químicos, es decir, a la estructura y composición atómica de los metales en sí.
Si recordamos un poco nuestras clases de química elemental, nos daremos cuenta que un elemento químico cualquiera, como cada uno de los metales, está constituido por átomos idénticos y únicos en el Universo. Cada átomo está formado de un núcleo conteniendo partículas elementales como los protones y los neutrones. Rodeando a este núcleo se encuentran los electrones, unidades elementales de carga eléctrica negativa que forman una nube que envuelve al núcleo atómico. Los electrones en un átomo ocupan niveles energéticos específicos y de diverso orden. Es decir, de acuerdo a la posición de un electrón en el átomo, aquel poseerá mayor o menor energía. Dependiendo de la complejidad del átomo, esto es, del número de electrones, las capas y orbitales se irán llenando de éstos en diversos niveles y por lo tanto sus energías serán diferentes de átomo a átomo. Esta estructura está reflejada en el arreglo encontrado en la tabla periódica de los elementos.
Las energías de un átomo surgen pues de energías de correlación debidas principalmente a interacciones entre electrones afectando su campo electrostático. Esto es muy importante en la química de los elementos ya que las energías que se involucran están directamente relacionadas con las reactividades químicas o con las diferentes tendencias para cambiar de un estado libre, como en el caso de un metal, a un estado de ión metálico, en donde el átomo ha cedido uno o más electrones.
Para ilustrar esta diferencia, tomemos de la naturaleza el ejemplo de los llamados metales nobles tales como el oro y el platino, que se encuentran usualmente como metales libres en la tierra y no como compuestos. Por otro lado, existen los llamados metales activos o base tales como el sodio, aluminio y magnesio, los cuales jamás se podrán encontrar como metales libres en nuestra atmósfera terrestre, sino como compuestos. Existen ocasiones en que ciertos metales con actividades intermedias a los anteriormente citados, pueden encontrarse en la tierra como elementos libres. Ejemplo de tales metales son el cobre, la plata y el hierro.
Los metales más "nobles", tales como el oro y el platino, son los menos "activos" y por tanto presentan la mayor resistencia a la corrosión. A estos metales se les refiere como metales relativamente catódicos. En cambio los metales menos "nobles" como el caso del aluminio y el magnesio, son más ''activos'' y poseen una resistencia menor a la corrosión y se les conoce como metales que son relativamente anódicos.
Una reacción de corrosión puede expresarse parcialmente por la ionización de
un metal, es decir, el proceso por el cual un átomo metálico pierde electrones
y queda cargado con un exceso de cargas positivas (iguales a las cargas negativas
de los electrones que se perdieron). Dicha entidad cargada constituye un ión
positivo o catión. Así pues:
siendo M un metal de valencia n, M+n su forma iónica y e el número de electrones cedidos (figura 6).
Ahora bien, si nosotros deseamos conocer la posibilidad de que una reacción
de corrosión ocurra espontáneamente bajo ciertas condiciones reales dadas, tendremos
forzosamente que estudiar primeramente cuales serían los cambios energéticos
asociados con la reacción. Esto es, ver la magnitud de la energía que el metal
poseía inicialmente antes de corroerse y luego ver la energía que poseen finalmente
los productos de esa corrosión. Esto es precisamente a lo que se dedica la termodinámica
de la corrosión. La termodinámica nos puede indicar una posibilidad de reacción,
pero jamás nos dirá nada acerca de la velocidad con que se llevará a cabo, si
es que la reacción es posible. Esto se decide por otros factores ajenos a la
termodinámica, factores propios de lo que conocemos como cinética.
Con el fin de que la distinción entre la aplicación de la termodinámica y la cinética en un proceso de corrosión le quede al lector lo suficientemente claro, vamos a usar una analogía muy útil. Supongamos que una persona ha subido a lo alto de un tobogán y está a punto de deslizarse. Por su posición, esta persona posee una energía dada (energía potencial) para hacer un trabajo. Al deslizarse hacia abajo, su energía cambia, disipándose gradualmente; al llegar al suelo nuestra persona ha adquirido otra energía debido a su posición, la cual es un poco menor que la inicial, cuando se encontraba arriba. La diferencia entre ambas energías fue lo que propició la caída. Se dice pues que hubo un cambio de energía durante el proceso de bajada. Así también cuando hablamos de una reacción de corrosión, estamos hablando de la energía que el metal y otros reactivos tienen al inicio de la reacción y la energía que poseen los productos finales de tal reacción. Habrá también un cambio en energía del estado inicial y del estado final. La magnitud y signo de tal diferencia nos indicarán la mayor o menor tendencia a que el proceso ocurra. Para cualquier proceso, el cambio de energía se calcula de acuerdo a:
Diferencia de energía = Energía final - Energía inicial
|
Cambio de energía
|
= |
Energía libre
|
—
|
Energía libre
|
|
libre de reacción
|
de los productos
|
de los reactivos
|
Cuanto mayor sea dicho cambio de energía, mayor será la tendencia a que el suceso
ocurra. Si el signo del balance es negativo implica que se está pasando de un
estado de mayor energía a otro de menor. A este tipo de proceso se le llama
espontáneo y ocurre en la naturaleza por sí solo, disipando energía. Ejemplo,
la caída de una piedra desde un puente, la degradación de hierro en un medio
ácido, etc. Esto es lo que estudia la termodinámica, cambios de energía, tendencias
a que el proceso ocurra o no, sin decirnos nada acerca de la rapidez con que
el proceso o la reacción se llevan a cabo.
La cinética nos da por otro lado los factores que afectan la velocidad del proceso. En el ejemplo del tobogán podemos decir que aun sabiendo que existía una probabilidad energética muy alta para que una persona dada resbalara desde la parte más alta hasta el piso, habían factores que debían tomarse en cuenta para darnos una idea de cuan lento podría resultar el suceso. Por ejemplo, debimos considerar si el ángulo de deslizamiento del tobogán era adecuado o si estaba muy empinado o muy tendido, haciendo más fácil o más difícil el movimiento; también había que tomar en cuenta la superficie del tobogán, es decir, su rugosidad, la fricción causada entre el cuerpo que resbala y el canal del tobogán, la sinuosidad misma del camino a recorrer hacia abajo, etc. Hay muchos más factores que podríamos considerar que obstaculizaban o facilitaban el proceso mismo, haciéndolo más o menos difícil. Así pasa con la cinética de una reacción electroquímica como lo es el caso de la corrosión, en donde siempre habrá una dificultad dada a vencer para que el proceso ocurra, a pesar de que la termodinámica nos diga que la reacción es espontánea y que ocurrirá desde un punto de vista energético.
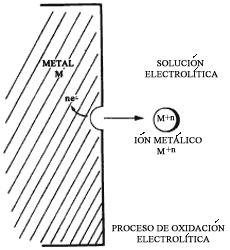
Cuando dos metales diferentes, que tienen, por lo tanto reactividades diferentes, son sumergidos en una misma solución conductora, a la cual llamamos electrolito, y son conectados eléctricamente entre sí, tendremos un flujo de electrones del metal más activo o anódico hacia el metal más noble o catódico, dejando al material anódico con una deficiencia de electrones (figura 7). Esto trae como consecuencia que el metal más activo o ánodo se disuelva, es decir, se corroa.
El lector recordará del capítulo anterior que a este sistema se le denomina
una pila de corrosión, la cual está caracterizada por tener los componentes
básicos para que un proceso de corrosión ocurra, que son: dos fases eléctricamente
conductoras (el hierro y el cobre), un medio acuoso en donde existan iones conductores
de la corriente eléctrica (la solución de cloruro de sodio) y, para cerrar el
circuito, un conductor eléctrico que una a las dos fases conductoras (el alambre
de cobre). Es bien sabido que si uno de estos componentes falla, la corrosión
no tendrá lugar. La corrosión ocurre en lo fundamental como se ha descrito anteriormente:
una reacción electroquímica (porque hay flujo de electrones y también transformaciones
químicas) que se lleva a cabo simultáneamente en zonas catódicas y anódicas.
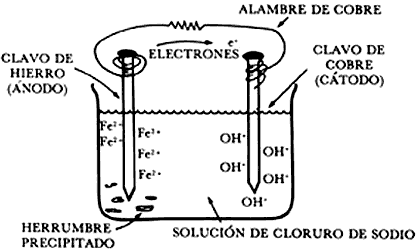
Dejemos que la mente nos ayude a construir una imagen de lo que realmente sucede a nivel atómico durante un proceso de corrosión. Vayamos a la zona anódica de la reacción, es decir, a la zona en donde el hierro se esta destruyendo gradualmente (figuras 7, 8(a)).
En ese lugar, el material anódico (el hierro) cede electrones al circuito eléctrico externo (alambre de cobre) debido a la diferencia de potencial creado por el par hierro cobre. Como habíamos explicado anteriormente, cada metal (hierro y cobre) posee una estructura atómica diferente y, como consecuencia de ello, una reactividad eléctrica local a través de la interfase entre el metal y el medio que lo rodea. Al entrar en contacto el hierro y el cobre se crea pues una diferencia de potencial eléctrico entre los dos metales, que hace que se mueva un flujo de electrones entre ellos.
Las partículas metálicas sobre la superficie del clavo de hierro (que en un principio eran neutras) ceden electrones y se convierten en átomos cargados positivamente (iones Fe2+) los cuales al interaccionar con las moléculas de agua pasan a la solución como especies solubles hidratadas. Reacciones químicas posteriores harán que estos iones Fe2+se transformen en el óxido rojizo o herrumbre, tan familiar para nosotros.
Pero ¿qué ocurre con los electrones cedidos por los átomos de hierro que pasan la disolución?, ¿hacia dónde van a parar? Anteriormente ya habíamos planteado esta pregunta y vimos que la respuesta era simple. Debido a la diferencia de potencial creada, los electrones viajan a lo largo del circuito conductor externo hacia el cátodo (el clavo de cobre, figuras 7, 8(b)). Ahí, los electrones en exceso, presentes sobre la superficie catódica, se combinan con otras especies en solución con el fin de balancear la reacción química de corrosión. El oxígeno del aire disuelto en la solución es una de las especies que tienen afinidad por los electrones y que en combinación con el agua se transforma en especies oxhidrilo (OH—). Así pues, el metal anódico se desintegra, mientras que el material catódico, el clavo de cobre en el ejemplo, permanece casi sin ser afectado.
Debe quedar claro que un proceso de corrosión involucra tanto la reacción anódica como la catódica, y que si una de las dos reacciones falla, el proceso de corrosión se detiene.
A la actividad electroquímica de esta naturaleza se le conoce como acción galvánica, base de cualquier reacción de corrosión. La acción galvánica constituye también el principio de las llamadas pilas "secas" que todos conocemos. El lector empieza a sospechar que un metal que se corroe es análogo a un sistema capaz de producir energía electroquímica, tal como lo es una pila. En estos sistemas también se establece la formación de un par galvánico, una diferencia de potencial producida por la presencia de dos fases diferentes y un electrolito entre ambas fases. Supongamos que establecemos la celda galvánica mostrada en la figura 9.
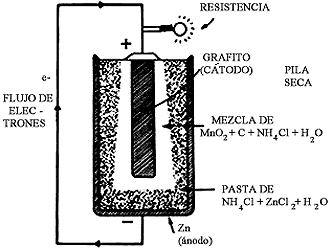
Siendo el cinc más activo que el grafito, aquél se disolverá preferencialmente
constituyéndose en el ánodo de la pila. Los electrones fluirán externamente
a través de una resistencia. Para mantener esta corriente eléctrica, el cinc
continuará disolviéndose y la barra de grafito sosteniendo reacciones catódicas
con la pasta electrolítica. De modo que el par cinc-grafito es una celda productora
de energía y se comporta muy similarmente a la celda galvánica de hierro-cobre
mostrada en la figura 7, en donde suceden procesos anódicos y catódicos similares.
Por lo dicho anteriormente, el lector podría pensar correctamente que entonces cualquier par galvánico es capaz de crear energía en forma de una diferencia de potencial capaz de mover electrones y, aún más, que la corrosión es un proceso que produce energía. Correcto. El lector está en lo cierto. Un par galvánico, como el que está presente en cualquier proceso de corrosión, es capaz de generar una cierta energía. Sin embargo, y ésto hay que admitirlo tal cual es, la energía generada por la corrosión no puede ser recuperada o usada. Se desperdicia sin que podamos hacer algo. Esto debido a que los procesos de corrosión ocurren a niveles microscópicos, en donde los pares galvánicos son sistemas tan locales que inclusive el conductor eléctrico externo, necesario en la constitución de una pila de corrosión, es la misma estructura metálica que se corroe. Aquí radica la diferencia entre un sistema productor de energía, la cual podemos extraer y usar (en una linterna, en el encendido del motor de un coche, etc), y un proceso de corrosión, análoga en sus reacciones, pero donde la energía no puede ser usada y es, en cambio, desperdiciada
Hagamos referencia a un ejemplo de corrosión muy común para todos nosotros: el deterioro del acero en la atmósfera. Esto lo vemos muy seguido en las varillas de acero usadas en la industria de la construcción, en tuberías, en carrocerías de coches que no están protegidas por pintura, etc. En primer lugar, tendremos que decir que el acero es una aleación de hierro y carbono (contenido máximo de 2% en peso) que presenta diferentes fases, producto de la historia metalúrgica del acero, tal como el método usado en su fabricación, la concentración de carbono, la presencia de aleantes, la refinación del acero y su tratamiento térmico, el cual es necesario para imprimirle ciertas propiedades mecánicas.
A nivel microscópico, una sección de la pieza de acero puede presentar la microestructura típica que se representa esquemáticamente en la figura 10. Se observan principalmente dos fases. Una fase rica en hierro que los metalurgistas llaman ferrita y otra fase conteniendo carburos de hierro en una matriz de ferrita. A esta última fase se le denomina perlita. Puesto que son dos fases diferentes y ambas son conductoras, es fácil establecer un par galvánico con la presencia de un electrolito.
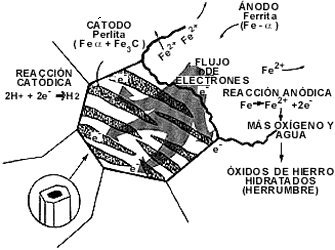
El electrolito lo constituirá la humedad que existe en la atmósfera, la cual
forma películas muy finas y casi imperceptibles sobre la superficie metálica.
Esta película de agua acumulará y concentrará ciertos elementos e impurezas
que están presentes en el aire, tales como el oxígeno, bióxido de carbono, anhídrido
sulfuroso, cloruros, etc., dando como resultado una solución muy conductora
y específicamente agresiva. En climas secos, con menos del 50% de humedad relativa,
la oxidación del hierro es casi despreciable. Así pues, la pila de corrosión
queda constituida, la conexión eléctrica es la misma pieza metálica y el electrolito
formado sobre la estructura baña a ambas fases. La ferrita es siempre más anódica
(activa) que la fase perlítica (noble). La corrosión se produce a nivel de microceldas;
millones de ellas están distribuidas a lo largo y a lo ancho de la pieza de
acero. El mecanismo heterogéneo de la corrosión tiende a fijar las reacciones
anódicas y catódicas en sitios definidos de la superficie metálica. El lector
seguramente ha concluido que basta la presencia de heterogeneidades, aún dentro
de una sola fase metálica, para que la corrosión se presente, existiendo, claro
está, la presencia de un electrolito. Estas heterogeneidades pueden ser de un
tipo o de otro, por ejemplo, diferentes fases metalúrgicas de una aleación,
tales como la perlita y la ferrita, producto de muchas variables metalúrgicas
como se mencionó anteriormente, la presencia de impurezas, una distribución
no uniforme de esfuerzos en el metal, arreglos diferentes en la red cristalina,
etc.
De cualquier manera, la lámina de acero se degradará con el tiempo. Fue preciso un gran flujo de electrones durante el proceso de corrosión, pero desgraciadamente en sucesos como estos, no se puede aprovechar nada de esa energía disipada.
CONVERSIÓN DE LA ENERGÍA DE UNA REACCIÓN QUÍMICA EN ENERGÍA ELÉCTRICA
Hemos estado hablando de energías eléctricas disipadas que provienen de una transformación de las energías originadas en una creación química. El objetivo de esta sección es el de correlacionar la energía química de una reacción con la energía eléctrica, en forma de diferencia de potencial eléctrico presente en una reacción electroquímica como lo es la de corrosión.
Cuando una reacción química sucede espontáneamente, libera energía. Este tipo
de energía puede ser empleada parcialmente para efectuar algún tipo de trabajo,
y es a lo que los termodinámicos denominan energía libre de una reacción y la
representan como DG. Si el cambio de energía
libre en una reacción es igual al trabajo reversible total que se puede realizar,
esto se puede representar de la siguiente manera:
en donde — DG es el cambio de energía libre de una reacción espontánea y W es cualquier tipo de trabajo, por ejemplo eléctrico, de expansión (en motores de combustión interna), gravitacional, etcétera.
Cuando nos referimos a una reacción de corrosión, tal como la representada en la figura 10, teníamos que por el lado anódico el hierro se disolvía a través de cantidades equivalentes, químicamente hablando, a las generadas en la cátodo. En este último los protones tomaban los electrones dejados por el hierro para transformarse en una molécula de gas hidrógeno.
La reacción global era de hecho:
Habiendo ocurrido un cambio en la energía libre asociada con esta reacción. Pero también ha ocurrido algo más, esto es, el transporte de dos cargas eléctricas debido a la diferencia de potencial existente entre las dos fases diferentes que constituyen el sistema de corrosión (ferrita y perlita en el sistema acero). A esta diferencia de potencial se le llama potencial de celda. Vamos a suponer que esta diferencia de potencial es igual a la diferencia de los potenciales termodinámicos en equilibrio de ambas fases.
Volviendo al transporte de las cargas, es obvio que tuvo que existir un trabajo para transportarlas de una fase a otra. A este trabajo se le llama trabajo eléctrico y se le define como la carga total transportada (dos electrones por molécula de hidrógeno formada) multiplicada por la diferencia de potencial existente entre las dos fases, es decir, DEº. Se tiene entonces que:
Aquí, el número de electrones transportados, n, es afectado por F, una constante
de transformación electroquímica llamada Faraday y usada para homogeneizar unidades.
Como no se obtiene otro tipo de trabajo de una reacción de corrosión, resulta
ser que la cantidad máxima de trabajo útil que se puede obtener de una reacción
química se ha transformado en energía eléctrica, al transportar las cargas entre
las dos fases, es decir:
el cambio de energía libre de una reacción química es directamente proporcional al potencial de celda generado. Este concepto es muy importante en el tratamiento de pilas "secas" o "húmedas" y en fuentes de energía alterna como lo son las celdas de combustible.
Las diferentes tendencias de los metales a corroerse: la Serie Galvánica
A través de mediciones de laboratorio, es posible construir un cuadro de metales o aleaciones de acuerdo a su tendencia relativa a corroerse. Estas mediciones están basadas en el principio de la acción galvánica que existe entre dos metales cuando se sumergen en un electrolito común. Todo lo que necesitaríamos para hacer tal cuadro sería medir la diferencia de potencial entre pares de distintos metales y aleaciones, teniendo una solución conductora común que bien podría ser agua de mar. Los metales se irían ordenando de acuerdo a su mayor o menor tendencia a corroerse. A tal enumeración se le conoce como Serie Galvánica, en donde el arreglo de los metales dependerá del electrolito escogido. La tabla siguiente muestra una Serie Galvánica para agua de mar.
|
Extremo noble
|
oro
|
|
acero inoxidable (pasivo)
|
|
|
níquel (pasivo)
|
|
|
cobre
|
|
|
bronce al aluminio
|
|
|
níquel (activo)
|
|
|
latón naval
|
|
|
estaño
|
|
|
acero inoxidable (activo)
|
|
|
hierro forjado
|
|
|
aluminio
|
|
|
Extremo base
|
cinc
|
|
magnesio
|
|
Por experiencia, se ha observado que aquellos metales que poseen un potencial
más positivo no sufren tanto por la corrosión y se les conoce como metales nobles.
Ejemplo: el oro, el acero inoxidable pasivo, el cobre, etc. Por otro lado, sabemos
que aquellos metales con potenciales más negativos se corroen rápidamente y
se les conoce como metales base. Como ejemplo tenemos al zinc, al magnesio y
al aluminio.
El lector observará en esta Serie que algunos metales o aleaciones aparecen dos veces, dependiendo de la condición en que se encuentra su superficie, es decir, activa o pasiva. Brevemente, podemos decir que el término pasivo se refiere a una superficie metálica que contiene alguna película de óxido protector. El término activo se refiere a la superficie metálica desprovista de tal película. Las superficies activas siempre están asociadas con potenciales más activos o base que las superficies pasivas.
Sin embargo, es importante establecer que la información contenida en estas series carece de una base científica precisa. Sus posiciones relativas dependen de una concentración específica del electrolito y también de la temperatura. Durante aplicaciones de campo, la tendencia de un metal o aleación a corroerse puede alterarse si es que hay variaciones en el medio ambiente. Si el electrolito cambia o al menos su concentración lo hace, tendríamos seguramente un nuevo arreglo de los metales en el cuadro ofrecido.
Serie electromotriz de potenciales estándar de electrodo
Un intento más para poner todo lo anterior en una base más cuantitativa, lo constituye el diseño de una escala en donde los diversos metales se colocaban en soluciones que contenían sus mismas sales. Las condiciones de medida son muy rígidas, especificando precisamente valores idénticos de temperatura, de presión y de concentración de los electrolitos. Al conjunto de estas condiciones se les conoce como condiciones estándar. En la práctica cada metal se observa en equilibrio en una solución de sus propios iones a una concentración de un gramo ión por litro de solución, a una temperatura estándar de 25ºC y a una presión atmosférica estándar. Así por ejemplo, el hierro sería medido bajo esas condiciones en una solución que contuviera iones hierro y el cobre en una solución con iones cobre, etc.
Cuando un metal M1, en contacto con sus sales en disolución M1n+ es medido en condiciones estándar contra otro metal diferente M2 haciendo de igual manera contacto con sus sales M2n+, la lectura de la diferencia de potencial resultante entre los dos sistemas será irrelevante si es que desconocemos los valores absolutos de cada una de las intercaras metal/solución. De hecho, es posible conocer el valor absoluto del potencial de cualquier interfase metal/solución, ya que en el mero intento de efectuar una medición con cualquier instrumento, siempre se tendrá que emplear una segunda terminal para cerrar el circuito de medida, introduciendo así una segunda interfase, cuyo potencial absoluto también se desconoce. Debemos de estar conscientes, pues, de que nunca podremos saber el potencial "verdadero" o absoluto de un metal sumergido en una solución de sus propias sales, por ejemplo, cobre en contacto con una solución saturada de sulfato de cobre. Ante esta situación, se pensó en seleccionar arbitrariamente alguna interfase como patrón de referencia contra la cual se pudieran medir relativamente todos los demás potenciales de electrodo. Se seleccionó para ello la reacción de equilibrio de hidrógeno: 1/2 H2=H+ e, llevada a cabo sobre un electrodo de platino y bajo las mismas condiciones estándares. A esta configuración de referencia se la conoce como electrodo estándar de hidrógeno y se le asigna un valor de 0.000 volts. A partir de esta convención es posible pues, medir potenciales estándar de electrodo de cualquier metal... relativos a la reacción de hidrógeno e indicarlos de esta manera: EºH.
A la lista de metales en equilibrio con sus propios iones junto con sus potenciales
estándares obtenidos de tales mediciones, se les llama la Serie de Fuerza Electromotriz,
de la cual se ofrece una versión muy reducida en la tabla siguiente.
| Reacción
en Equilibrio
NOBLE |
E°H (volts) |
||
| Au+2
1/2 O2
Pt+2
Ag+1
Cu+2
2H+
Ni+2
Fe+2
Cr+3
Zn+2
Al+3 |
+ 2e
+ 2H+
+ 2e
+ 1e
+ 2e
+ 2e
+ 2e
+ 2e
+ 3e
+ 2e
+ 3e |
= Au
+ 2e = H2O
= Pt
= Ag
= Cu
= H2
= Ni
= Fe
= Cr
= Zn
= Al |
+ 1.7
+ 1.23
+ 1.20
+ 0.80
+ 0.34
0.00 (por definición)
- 0.13
- 0.44
- 0.70
- 0.76
- 1.66
|
| BASE | |||
La información contenida en esta serie electromotriz, representa simplemente
la tendencia termodinámica (el potencial) de los varios sistemas ahí en lista
destinados a corroerse. Cuanto más negativo sea el valor de potencial E°H
mayor será la tendencia a la corrosión. La serie electromotriz nos
puede explicar cuantitativamente, por ejemplo, la tendencia y el porqué de una
adherencia de cobre metálico sobre un clavo de acero cuando sumergimos el clavo
en una solución de sulfato de cobre. En este caso la fase noble la constituye
el cobre que se encuentra en disolución y la activa o base el clavo de acero.
La Serie nos indica que existe una diferencia de potencial, bajo condiciones
estándar, entre el cobre y el hierro de 0.78 volts, siendo el hierro el metal
con mayor tendencia a ionizarse. De esta manera el cobre que está en solución
como iones cobre, tomará los electrones liberados al formarse los iones hierro
y pasará a depositarse como metal sobre el clavo. Lo que ocurrió aquí, una vez
más, fue la presencia de un par galvánico.
Sin embargo, al ponernos a reflexionar sobre ejemplos prácticos de corrosión, casos que suceden a diario, nos damos cuenta de que muchas estructuras se corroen o fallan por corrosión en la ausencia de otro metal más noble que complete el par galvánico de la pila de corrosión. Por ejemplo, ¿cual fue la reacción catódica o de reducción, en el ejemplo de la figura 10 para la corrosión de una pieza de acero que cerrara el balance galvánico? ¿Existían iones metálicos más nobles en la solución agresiva? La respuesta es negativa. No había tales iones metálicos más nobles, pero si reacciones tales como la reducción del oxígeno y de hidrógeno iónico, cuyo equilibrio, según nos dice la Serie de Fuerza Electromotriz es más noble que el hierro. Entonces podemos tomar estas reacciones como si se trataran de reacciones de "metales nobles" que en unión con un metal más activo desencadenan el proceso de corrosión.
Así entonces, haciendo esta consideración, podemos decir que todos los metales que se encuentran por debajo de reacción de reducción de hidrógeno se disolverán siempre que estén en contacto con un medio que contenga H+, tal como el agua o las soluciones ácidas. Así, tanto el hierro como el cinc se corroerán totalmente en soluciones acuosas o ácidas. En cambio, el cobre permanecerá inmune a la reacción de hidrógeno, pues de acuerdo a su posición en la Serie, esta última reacción es más activa que el mismo cobre. En cambio, el cobre es más activo y susceptible a disolverse en presencia de la reacción de reducción de oxígeno. Por ejemplo, es muy común la corrosión del cobre en contacto con agua aereada, es decir, agua que contenga oxígeno disuelto (ver figura 12). Sólo el oro permanece intacto ante soluciones que contienen protones H+ u oxígeno atmosférico, ya que su equilibrio es más noble que los de estas reacciones.
La serie de fuerza alectromotriz posee severas limitaciones a pesar de toda esa cantidad de información termodinámica tan interesante. La serie no considera el efecto que tiene una película de óxido presente en la superficie de un metal, en el potencial de equilibrio de ese sistema, siendo así que estas situaciones prácticas son las que a un ingeniero de planta le gustaría saber. Por ejemplo, la serie considera que tanto el aluminio como el cromo son sumamente reactivos (-1.66 y -0.70 volts respectivamente) y sin embargo, nosotros sabemos que el aluminio, tal como lo conocemos, es muy resistente a la corrosión en condiciones normales y que al cromo incluso se le usa como elemento de aleación en los aceros para imprimirles mayor resistencia a la corrosión. Lo que sucede es que la Serie no considera la condición oxidada tanto del aluminio (A12O3) como del cromo (Cr2O3), los cuales son así excepcionalmente resistentes a la corrosión; la serie sólo considera sus estados activos, es decir, no pasivos.
Otras reacciones que la Serie no toma en cuenta son reacciones muy comunes
entre un metal y un medio acuoso. Tomemos por ejemplo las reacciones siguientes:
(a) Fe + H2O = Fe (OH)3 + 3H+
+ 3e-
(b) Fe+3 + 3 H2O = Fe (OH)3
=3 H+
Aquí (a) nos indica la reacción que sufre el hierro en contacto con el agua para formar el hidróxido férrico. El equilibrio de esta reacción ya no depende solamente de un potencial eléctrico que haga mover esos tres electrones por mol de Fe(OH)3 formado, sino también del pH, es decir, de la acidez del medio, de la concentración de iones H+. (b) no depende del potencial sino sólo del pH del medio. Aquí no hay transferencia de electrones, es una reacción química, no electroquímica. Ambas reacciones y muchas otras son importantes para saber el estado en que se encontrará una estructura de acero, dependiendo del potencial y de la acidez del medio, por ejemplo, si habrá o no formación de herrumbre.
Si se contara con los equilibrios de todas las reacciones posibles entre un metal y el agua, nos podríamos dar una mejor idea de la tendencia que poseería ese metal ante un conjunto dado de condiciones de potencial y de pH, es decir, podríamos decir si hay en el metal tendencia a formar óxidos o hidróxidos, si tenderá a disolverse completamente o si, bajo esas condiciones, el metal permanecerá intacto.
Hace algunas décadas, el investigador belga Marcel Pourbaix no sólo obtuvo esos equilibrios sino que los representó gráficamente como función del potencial y del pH a través de unos diagramas que llevan su nombre. En estos diagramas, los equilibrios existentes entre un metal y agua a 25ºC son representados por líneas que dependen del potencial, del pH o de ambos, delimitando así zonas termodinámicamente estables en donde el metal existe en alguna de sus formas (disuelto, como óxido, o como hidróxido, como metal, etc.).
La figura 11 (a) nos muestra el diagrama simplificado para el hierro en contacto con agua. Un aspecto importante de los Diagramas de Pourbaix es que contienen una división natural del campo gráfico en tres regiones, las cuales pueden ser clasificadas de acuerdo a su conducta de corrosión en: pasividad, corrosión e inmunidad, figura 11(b). La zona de pasividad, se aplica para cuando el metal posee películas oxidadas o de hidróxidos sobre su superficie que inhiben la corrosión. En la zona de corrosión, el metal se disuelve activamente, siendo los productos de corrosión solubles. En cambio en la zona de inmunidad, el metal se encuentra perfectamente preservado y estable bajo ciertas condiciones muy especiales de potencial y de pH.
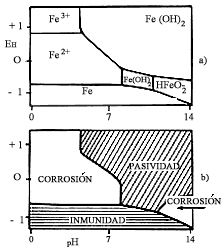
Antes de cerrar esta sección dedicada a la termodinámica en los procesos de
corrosión, queremos nuevamente recordar a nuestro amable lector que en el estudio
termodinámico de un proceso de corrosión no se incluyen los datos cinéticos.
Esto significa que la velocidad del proceso no se toma en cuenta, solo la tendencia
a que la corrosión ocurra.
Cinética de un proceso de corrosión
Establecimos en la sección anterior que la termodinámica aplicada a procesos de corrosión puede ser usada a través de reacciones en equilibrio para determinar si el proceso puede o no puede ocurrir. Si se demuestra que la reacción es posible, la termodinámica no dará, sin embargo, ninguna idea acerca de la velocidad de la reacción. Para predecir la velocidad a la cual el metal se va a corroer necesitamos incluir factores cinéticos. A final de cuentas, lo que buscaremos de la cinética es que nos diga qué cantidad de metal por unidad de tiempo se está disolviendo en un medio dado, cuando este sistema esté desplazado de una situación de equilibrio. Para introducirnos al mundo de la cinética de corrosión debemos olvidarnos por un momento de la corrosión y pensar en un metal que no se corroe cuando se le sumerge en cierto electrolito.
Pensemos que ese metal sea el cobre y que esté sumergido en una solución conteniendo
iones cobre. La solución no contiene oxígeno disuelto. Bajo estas condiciones
nuestra pieza de cobre no se corroerá. Esto se debe fundamentalmente a que el
cobre es mucho más noble que la reacción de desprendimiento de hidrógeno según
lo indica la Tabla de la Fuerza Electromotriz, ECu > EH.
Aparte del hidrógeno iónico no existe otro reactivo más catódico en ese medio,
por lo tanto decimos que el cobre establece un equilibrio del tipo
que ocurre precisamente al potencial relativo estándar EºCu/Cu2+ establecido anteriormente.
Al decir que la pieza de cobre no se corroe cuando está en contacto con una disolución de sus propios iones, queremos expresar que no hay una transformación química neta. Sin embargo, debemos dejar muy claramente establecido que aun cuando el sistema se encuentre en equilibrio, éste no es estático sino dinámico. Al potencial Eº, existen reacciones de oxidación por las cuales se disuelve cobre, y también reacciones de reducción en donde cobre en solución se deposita como metal. Estas reacciones ocurren de continuo y simultáneamente, siendo iguales en la magnitud de la carga transferida, aunque en sentido contrario. La velocidad con que se disuelve el cobre de la pieza es igual a la velocidad con que el cobre en solución se deposita. En resumen, en el equilibrio dinámico Eº, no existe flujo de electrones ni, por lo tanto, transformación química netos. Figura 12.
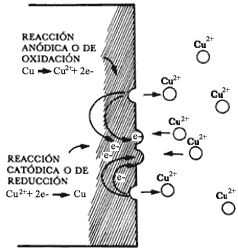
Es como si pensaramos en una cantidad de personas que entran y salen por la puerta de un gran almacén. En una situación de equilibrio diríamos que el mismo número de personas que entra al almacén sale de él. Por lo tanto, no existe cambio alguno en el número de personas del lado de la calle ni dentro del almacén; parecería como si nunca hubiera habido un movimiento. En electroquímica, a este flujo de electrones generados en la reacción de disolución (oxidación), el cual es igual al flujo de electrones consumidos en la reacción de reducción, se le llama densidad de corriente de intercambio io, siendo una característica de un metal en equilibrio. Cada interfase tendrá una io característica y su magnitud refleja la facilidad que posee esa interfase metal/solución para soltar y aceptar electrones. Volviendo al ejemplo del almacén, podemos pensar en magnitudes diferentes de flujos opuestos de personas que entran y salen, pero que siempre mantendrán el mismo número dentro y fuera del almacén. Así por ejemplo, si el almacén tiene en un momento dado 1 000 personas y en la calle hay otras 5 000 para mantener esta relación bien pueden entrar al almacén 10 personas y salir simultáneamente otras 10. Pero bien podrían haber entrado 100 y haber salido también 100, manteniendo el mismo número de personas dentro y fuera. Tal vez un aumento grande en el flujo de entrada y salida del almacén se deba a que las puertas sean más anchas, es decir, que la facilidad de traspaso sea mayor. Lo mismo sucede con interfases electroquímicas, a mayor io, mayor facilidad de transferencia de carga, y viceversa.
Como lo mencionamos, cada interfase en equilibrio posee una io específica, cuya magnitud proviene de consideraciones cinéticas con más fundamento, tales como arreglos atómicos, enlaces electrónicos, etc, que no son consideradas en este libro. Sin embargo, la importancia de io en los procesos de corrosión será tratada más adelante en este capítulo.
Por supuesto, como las velocidades de oxidación y de reducción son iguales
en magnitud pero de signo opuesto, no habrá un flujo de electrones neto
y, por lo tanto, será imposible medir la io en un instrumento.
Si la corriente anódica (de oxidación) se representa por ![]() y la corriente catódica (de reducción) como
y la corriente catódica (de reducción) como ![]() ,
en el equilibrio
,
en el equilibrio
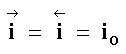
en donde io es, como ya lo dijimos, la densidad de corriente de intercambio.
Cualquier desviación que haya de la condición de equilibrio, desplazará el potencial del electrodo y entonces modificará las magnitudes de las velocidades anódicas y catódicas, produciéndose un flujo neto de electrones. Supongamos que nuestra pieza de cobre, inicialmente en equilibrio con sus propios iones, se conecta a una laminilla de platino sumergida en el mismo electrolito a través de una fuente de poder. Si asumimos que al manipular la fuente de poder podemos establecer una diferencia de potencial entre el cobre y el platino que haga fluir electrones entre estos metales, entonces se estarán modificando las condiciones de equilibrio reinantes en cada una de las interfases del cobre y del platino. Imaginemos que la diferencia de potencial impuesta por la fuente propicie que el cobre se disuelva, actuando éste como ánodo, pasando a solución como iones cobre Cu2+. Habrá entonces un flujo de electrones generados que viajarán hacia el platino (que actuará entonces como cátodo) a través de las conexiones eléctricas. El flujo de electrones ha propiciado que los potenciales de equilibrio de cada una de las fases se desvíen de su valor inicial. Esto es lo que precisamente significa el término polarización, el desplazamiento de un potencial de equilibrio hacia otro valor por medio de un flujo de corriente eléctrica. Un símil hidráulico muy útil en este caso es el ejemplo de los vasos comunicantes. Supongamos que se tienen dos vasos o recipientes llenos con agua a diferente nivel. Si dichos vasos están comunicados entre sí a través de una llave de paso, cuando ésta se abra y deje pasar el flujo, los niveles iniciales de los líquidos serán modificados (figura 13). La magnitud del cambio de nivel dependerá, en el símil hidráulico, de la velocidad del agua que fluye; algo parecido sucede con un proceso de corrosión en donde hay una diferencia de potencial eléctrico entre dos fases diferentes.
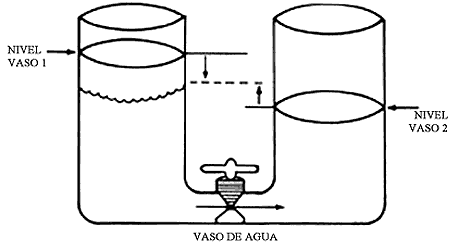
En la interfase cobre/solución se dejará sentir pues, un incremento en la velocidad de oxidación del cobre. Las reacciones de reducción en esa interfase aún existirán, aunque ahora comparativamente más pequeñas que las de oxidación. Las viejas condiciones de equilibrio han desaparecido para dar cabida a nuevas situaciones al no equilibrio. En la lámina de cobre, la magnitud de la corriente indicará el grado en que la velocidad de oxidación, por decir algo, excederá a la reducción. Es decir, nos dará una indicación de la corriente neta del proceso. La velocidad neta del proceso de oxidación o anódico estará dada por:
A principios de este siglo, un investigador llamado Tafel halló en forma experimental
que, a menudo, el flujo neto de corriente variaba linealmente con la magnitud
de la desviación que tiene el potencial de equilibrio, h,
a través de la relación:
h = a + b log i(neta)
en donde h se le conoce como sobrepotencial y se le define como
Es decir, h es la magnitud de la desviación del potencial electroquímico del sistema en observación (en este caso cobre) a partir de su valor de equilibrio original, ineta es la densidad de corriente neta anódica o catódica y a y b son constantes. A esa relación se le llama, en honor de dicho investigador relación de Tafel y es de suma importancia en el análisis cinético de un proceso de corrosión. El lector habrá notado seguramente que esta relación de Tafel es la de una ecuación de una recta, en donde la variable independiente es la corriente presentada en forma logarítmica y la variable dependiente es el sobrepotencial, figura 14.
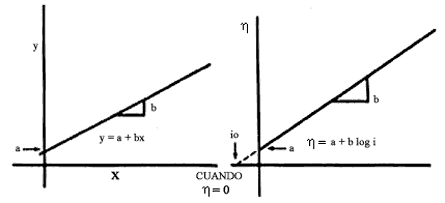
El valor de la constante "a" esta relacionado con la velocidad de las reacciónes anódicas y catódicas ( io) bajo condiciones de equilibrio, es decir, cuando el sobrepotencial h es cero. Cuando la desviación del potencial de equilibrio es positiva (+h) se dice que el proceso es anódico, o sea, el metal se oxida o disuelve. Si la desviación es negativa (— h), el potencial aplicado toma valores más negativos que el de equilibrio y el proceso es catódico, habiendo reacciones de reducción en la interfase metal/medio electrolítico.
La figura 15 representa gráficamente la relación de h contra log i para la reacción anódica de disolución (corrosión) metálica de la pieza de cobre considerada en el ejemplo cuando su potencial de equilibrio sufre una desviación en la dirección positiva (+h) (cuadrante superior derecho). Se observa que al incrementar la desviación en la dirección anódica, se estimula la velocidad de disolución de Cu (log i). En la misma figura se aprecia la conducta que sucedería en el caso de que la lámina de cobre hubiera sido alejada del equilibrio pero ahora en la dirección negativa o catódica (— h) (cuadrante interior izquierdo). Esto se realizaría simplemente cambiando la polaridad de las conexiones en la fuente de poder. La reacción catódica que se efectuaría, sería predominantemente la depositación de cobre como se indica. Ambos procesos giran alrededor del potencial de equilibrio.
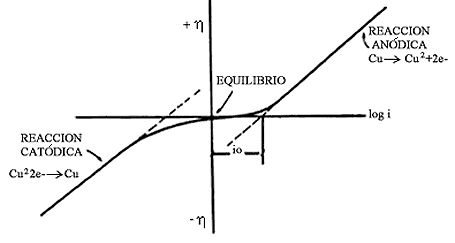
Algunas veces es difícil de interpretar la figura 15, especialmente cuando tratamos con la magnitud de io. Una manera más familiar y también fácil de ver esta misma relación es como la que se representa en la figura 16. Esta representación se obtiene al doblar el lado izquierdo sobre el derecho tal y como se muestra en el recuadro. A este tipo de diagramas se les conoce con el nombre de Diagramas de Evans.
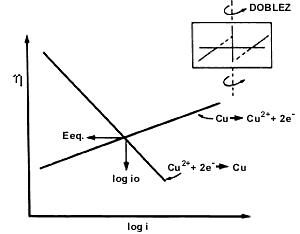
Un ejemplo sencillo de la Cinética de un Proceso de Corrosión
En contraste al caso del cobre, cuando un pedazo de zinc se pone en una solución acuosa deareada de sus propios iones, éste se corroe. Esto es debido a que el desprendimiento de hidrógeno es termodinámicamente posible (Ezn<EH). Debido a heterogeneidades presentes en la superficie del metal zinc, se establecen de inmediato zonas anódicas y zonas catódicas las cuales desarrollan entre sí diferencias de potencial, acarreando como consecuencia de ello un flujo neto de electrones de las zonas anódicas a las catódicas y con ello, el establecimiento de la corrosión. Así es que los dos sistemas electroquímicos presentes, uno basado en el equilibrio Zn/Zn2+ y el otro en el equilibrio H+/H2, buscan inicialmente mantener un equilibrio sobre la misma superficie del metal. Sin embargo, la corrosión que ocurre lo impide. Por un lado el zinc se disuelve y por el otro el hidrógeno gaseoso se desprende de zonas metálicas que funcionan como cátodos. Al haber corrosión, hay un flujo de corriente y por lo tanto ninguno de los sistemas electroquímicos está en equilibrio. De ahí se desprende que ambos sistemas se desvían de sus potenciales de equilibrio (se polarizan) y alcanzan un potencial común de electrodo en donde sucede que la corriente anódica de disolución (la velocidad con que se corroe el zinc) es igual a la corriente catódica (la velocidad con que se desprende el hidrógeno). El diagrama de Evans de la figura 17 muestra el análisis de este suceso.
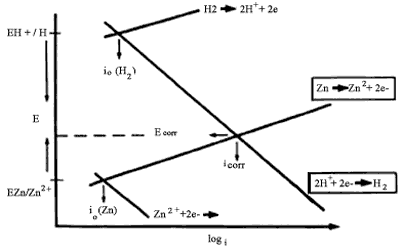
El potencial uniforme al que se llega sobre la superficie metálica se llama el potencial de corrosión (Ecorr). La velocidad de corrosión está dada por icorr, la cual puede ser convertida a pérdida de peso usando las Leyes de Faraday. El lector recordará de la sección anterior que aunque exista una disolución de zinc neta, ocurre también un desprendimiento neto de hidrógeno pero que no hay manera de poder medir o usar el flujo de corriente entre los pares galvánicos establecidos. Toda esta energía se desperdicia.
El ejemplo anterior pone de manifiesto, una vez más, que durante el proceso de corrosión de un metal se llevan a cabo simultáneamente reacciones anódicas y catódicas sobre la superficie de éste. En las zonas anódicas el metal se disuelve y en las zonas catódicas ocurren reacciones de reducción entre el metal y el medio, por ejemplo la reducción de H+. Ni las fases anódicas ni las catódicas se encuentran en su potencial de equilibrio (Ezn/Zn2+ y EH/H+ respectivamente). Según la figura 17 ambos potenciales de equilibrio son desplazados a un valor común, el potencial de corrosión, en el cual la velocidad de oxidación del metal (corrosión) es igual a la velocidad de reducción del protón H+. Por lo general, cuando medimos el potencial de un metal sumergido en un medio agresivo, contra un electrodo de referencia, lo que realmente medimos es el potencial de corrosión mixto, Ecorr.
La velocidad de corrosión de un proceso, icorr, puede tomar diversos valores dependiendo de varios factores que modifican su magnitud destacando:
a) la densidad de corriente de intercambio, io. La figura 18 demuestra este punto. La io para una reacción depende mucho de la superficie en donde se lleve a cabo. Así por ejemplo, es más fácil desprender hidrógeno sobre hierro (io2) que sobre zinc (io1). La velocidad de corrosión aumentará de icorr2 a icorr3
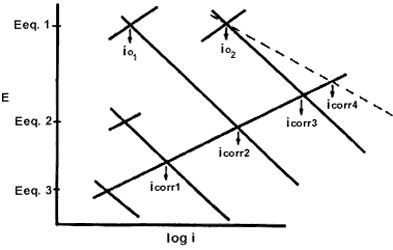
b) la fuerza motriz para llevar a cabo la corrosión, es decir, la diferencia
de potencial termodinámico del par galvánico. La figura 18 muestra que mientras
más grande sea la diferencia de potencial entre la reacción anódica y la catódica,
mayor será la velocidad de corrosión. Para (Eeq.2 - Eeq.3) corresponde una
icorr1. En cambio para (Eeq.1 - Eeq.3) la velocidad de corrosión
es icorr2.
c) un último factor lo constituye el valor de la pendiente de Tafel, la cual nos dice de la facilidad o dificultad de transferir cargas esta vez cuando una fase se aleja del equilibrio (las unidades de la pendiente son: volts/década de corriente). Haciendo referencia a la misma figura 18 observamos que al disminuir la magnitud de la pendiente para la reacción catódica (línea punteada), ésta se vuelve más fácil, aumentando por lo tanto la velocidad de corrosión de icorr3 a icorr4.
Cuando la conducta de Tafel se desvía
Como ya lo hemos mencionado en más de una ocasión, si un metal se corroe es porque hay reacciones anódicas y catódicas que se llevan a cabo simultáneamente sobre su superficie. En cada una de las áreas anódicas y catódicas el proceso global de una reacción es consumado a través de varias etapas. Así por ejemplo, en la reacción catódica de desprendimiento del gas hidrógeno, inicialmente el protón solvatado tuvo que migrar y difundirse hacia el electrodo, sufrir la transferencia de un electrón para convertirse en un átomo solitario de hidrógeno y esperar a que se repitiera la misma operación para que finalmente el par de átomos de hidrógeno se unieran formando una molécula de gas. Algo similar debió de ocurrir en el ánodo. La disolución de átomos metálicos y su conversión final en productos de corrosión característicos que permanecen sobre la superficie metálica o que se difunden lejos de ella, se habría llevado en varias etapas. Cada etapa tienen su propia rapidez. Siendo así, podemos decir que la velocidad global de un proceso considerado esta sujeta, o más bien depende de la etapa que se realice más lentamente. No debemos perder de vista que dentro del proceso anódico y catódico presentes en un metal que se corroe están las etapas de transferencia de cargas tanto de oxidación como de reducción respectivamente. Siendo así, nuestros lectores podrán pensar entonces que es posible que una sola etapa, sin importar si pertenece a la reacción anódica o a la catódica, sea la responsable de la velocidad global del proceso de corrosión de un metal en algún medio, si es que esa etapa es la más lenta de todas. Supongamos que la etapa más lenta sea la transferencia de un electrón del metal al protón, y que efectivamente sea este paso el más difícil. Entonces la reacción catódica se llevará a cabo con la máxima velocidad con que se transfiere el electrón al protón. Pero como en la corrosión la velocidad de la reacción anódica debe ser igual a la velocidad de la catódica, pues la velocidad de la reacción de corrosión es la misma que la velocidad de la etapa más lenta, es decir, la de la transferencia del electrón al protón.
Todo lo dicho anteriormente se justifica, ya que lo que se establece a continuación es importante. La conducta de Tafel se observa solamente cuando las velocidades de las reacciones anódicas o catódicas sean gobernadas por la etapa de transferencia de carga en la interfase metalelectrolito. Esto es, cuando esta transferencia sea el paso difícil o más lento.
Un ejemplo que clarifica mejor el concepto de un proceso realizado en multietapas, teniendo como etapa o paso limitante a la más lenta, la etapa de transferir cargas, o de activación como también se le conoce, es el ejemplo de un aficionado a la pesca. Esta persona sale temprano de su casa, toma su coche y se dirige al lago. Al llegar ahí nuestro pescador toma los arreos necesarios para la pesca, como son la caña, anzuelo, carnada, etc.; sube al bote y rema hasta el centro del lago en donde hay mayor cantidad de peces. Estando ahí lanza el anzuelo y espera que los peces piquen. Así, esta persona pasa el resto del día pescando tranquilamente. Si nos fijamos, el paso lento del proceso es la acción de los peces al picar; todos los demás pasos que el pescador tomó para efectuar el "proceso de pescar" son mucho más rápidos que el paso de obtener un pez. Así sucede también con los procesos regidos por la transferencia de carga (electrones) tanto en el ánodo como en el cátodo, de los cuales se dice que siguen la conducta de Tafel.
Sin embargo, las desviaciones a la conducta de Tafel surgen cuando la velocidad de la reacción pasa a ser controlada por una etapa más lenta en la secuencia del proceso. Imaginemos que nuestro pescador se encuentra cierto día con la novedad de que los peces no abundan en el lago. La lentitud del proceso de pescar ahora ya no será culpa de la facilidad o dificultad que tenían los peces de picar, sino de que no hay suficientes peces. Nuestro pescador tiene que esperar más tiempo para que un pez llegue al anzuelo que el que le tomaba para pescarlo. A este fenómeno similar en proceso de corrosión se le llama polarización por concentración y surge por deficiencia en el abastecimiento de reactivos que toman parte en una reacción electroquímica.
Polarización por concentración
La ecuación de Tafel sugiere que la densidad de corriente, i, aumentará continuamente
al aumentar el sobrepotencial , h figura 14.
De hecho, la velocidad de la reacción se verá limitada a menudo al aumentar
h, debido a la poca rapidez con que los reactivos
llegan a la superficie del electrodo o bien a la velocidad con que se difunden
hacia el seno de la solución los productos de la reacción. La velocidad ya no
es controlada por un paso lento de transferencia de carga y por consiguiente
habrá desviaciones a la conducta de Tafel. Esto implica que se necesitará un
mayor sobrepotencial que el que predice la relación de Tafel para poder seguir
sosteniendo una corriente dada. Si hacemos una distinción entre el sobrepotencial
por activación o transferencia (dado por Tafel) y el sobrepotencial por concentración
(debido a la escasez de reactivos o exceso de productos) entonces el sobrepotencial
total será dado por:
Esto es demostrado en la figura 19. Se observa que para condiciones críticas de abastecimiento, se llega a una corriente límite, valor que no se incrementa aún y cuando el h se incremente. Esto quiere decir que el proceso no puede ir más rápido que la velocidad que impone el transporte de las especies hacia el electrodo.
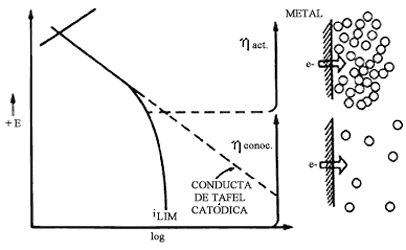
Las implicaciones de la polarización por concentración para un sistema que se
corroe son muy importantes. En casos prácticos, lo más común es que la concentración
por polarización afecte la reacción catódica debido, por ejemplo al abastecimiento
de H+ o de oxígeno disuelto. En la figura 20 se exponen dos
ejemplos, en uno de los cuales, (a), la iLim es grande y por
lo tanto la polarización por concentración no es importante. Esto es debido
a que la curva anódica intersecta a la catódica en la región de Tafel.
En el ejemplo (b) de la figura 20 se tiene que si iLim es pequeña las dos curvas se intersectan en la región controlada por difusión. Ahí iLim=icorr, y la velocidad de corrosión depende totalmente del transporte de reactivos catódicos hacia la superficie metálica. En este último caso el pobre abastecimiento del reactivo catódico, ya sea por una concentración baja de H+ o de oxígeno atmosférico disuelto en el medio agresivo, ayudarán a reducir la velocidad de un proceso de corrosión al controlar la reacción catódica.
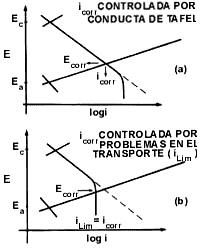
Figura 20.