IX. LOS RETROVIRUS: LA CLAVE DE LA ONCOGÉNESIS VIRAL
A PRINCIPIOS de los años sesenta, Howard Temin demostró que era
posible inducir con una alta frecuencia mutaciones en el genoma del virus del
sarcoma de Rous (RSV) presente en el interior de células infectadas por dicho
tipo de virus. Las mutaciones producidas en el genoma viral se manifestaban
como cambios en la morfología de las células infectadas y el genoma viral mutante
podía ser heredado en forma estable por la subsecuente progenie celular. Esta
observación condujo a pensar que la información genética del virus se encontraba
presente en una estructura de la célula hospedera capaz de ser heredada en forma
regular. Así nació la idea del "provirus" y la especulación de que este hipotético
provirus se encontraba integrado en el genoma de la célula hospedera. El principal
problema de esta hipótesis consistía en que hasta entonces no se conocía ningún
camino por el cual el ARN que constituye el material genético del
RSV pudiera ser convertido en ADN y de esta manera ser integrado
en el genoma de la célula hospedera. El llamado dogma central de la biología
molecular sostenía que la información genética solamente fluye en forma unidireccional:
ADN ![]()
ARN![]() Proteína. Sin embargo, Temin observó que ciertas substancias capaces de intercalarse
en la molécula de
Proteína. Sin embargo, Temin observó que ciertas substancias capaces de intercalarse
en la molécula de ADN y bloquear la síntesis de ARN
dirigida normalmente por el ADN, eran también capaces de bloquear
la síntesis de ARN viral en células infectadas por RSV.
Esta observación sugería en forma indirecta la posibilidad de que en este caso
la información genética fluía de ARN hacia ADN y de
nuevo hacia ARN ![]() Proteína.
Experimentos posteriores demostraron que era necesaria la síntesis previa de
un nuevo tipo de
Proteína.
Experimentos posteriores demostraron que era necesaria la síntesis previa de
un nuevo tipo de ADN en las células infectadas por RSV
para que se produjeran las nuevas partículas virales; por lo tanto, este nuevo
tipo de ADN era producido en las células como consecuencia de la
infección por RSV. Esta paradójica observación implicaba que de
alguna manera desconocida el ARN viral inducía la síntesis de un
cierto tipo de ADN, el cual a su vez era necesario para la futura
producción de nuevo ARN viral. Con base en estos resultados, Temin
póstuló en 1964 la hipótesis del provirus. Esta hipótesis propone que el ARN
que constituye el genoma del RSV infectante actúa como templado
para dirigir la síntesis de una molécula de ADN que es equivalente
al ARN viral original. Este nuevo ADN viral puede
integrarse en forma de provirus en el genoma (ADN) de la
célula hospedera donde subsecuentemente actuará como templado para la síntesis
de nuevo ARN viral que constituirá la progenie del virus infectante
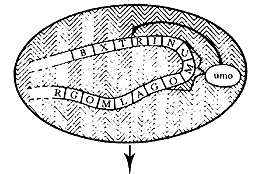
(figura IX.I).
Durante casi 6 años la hipótesis del provirus permaneció prácticamente ignorada
hasta que en 1970 Temin y Baltimore, trabajando por separado y en forma independiente,
demostraron la existencia de una actividad enzimática codificada por el genoma
del RSV; esta actividad es capaz de dirigir la síntesis de ADN
a partir de ARN viral. Esta nueva enzima fue bautizada como transcriptasa
inversa y constituyó el eslabón necesario para explicar el mecanismo de
transmisión de la información genética propuesto en la hipótesis del provirus.
La prueba formal de esta hipótesis fue obtenida por medio de experimentos de
hibridación de ácidos nucleicos en los cuales el ARN viral marcado
radiactivamente mostró un mayor índice de hibridación con el ADN
de células infectadas que con el ADN de células no infectadas por
RSV. Este resultado se debe a que el ADN de las células
infectadas contiene secuencias complementarias al ARN viral. Posteriormente,
experimentos realizados con ADN obtenido a partir de células infectadas
de antemano con RSV, mostraron que es posible transfectar;
o sea, introducir el ADN purificado en células no infectadas, mismas
que posteriormente darán origen a viriones completos de RSV a partir
de la información contenida en el ADN introducido a la célula.
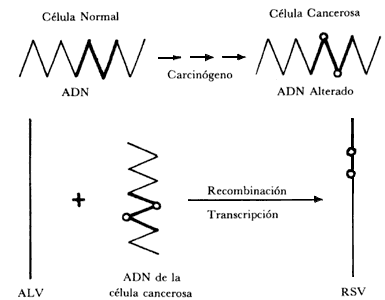
Figura IX.1. Hipótesis del protovirus para el origen de los genes cancerosos
y la generación de virus ARN oncogénicos. En el ejemplo ilustrado,
el virus del sarcoma de Rous (RSV) es visualizado como si se originara
a partir de un virus con bajo potencial oncogénico como el virus de la leucosis
aviaria (AVL). Las líneas zigzagueantes anchas indican ADN
involucrado en la transferencia de información desde el ADN y de
nuevo hacia ADN por medio de la enzima transcriptasa inversa. El
ARN del ALV incorpora el ARN transcrito
a partir del ADN anormal propio de una célula cancerosa y de esta
manera se convierte en el virus oncogénico RSV.
Temin propuso una extensión de la hipótesis del ADN proviral.
Esta nueva idea, conocida como la teoría del "protovirus", postula que cuando
menos una parte del genoma de los virus oncogénicos ha surgido como consecuencia
del proceso evolutivo a partir del ADN de células normales que
han sido alteradas por algún agente carcinogénico. La recombinación genética
entre el ADN viral y el ADN celular puede resultar
en carcinogénesis debido a la inserción del híbrido de ADN viral
y celular en un sitio inadecuado dentro del genoma de una céla hasta entonces
normal; esto puede propiciar la activación de genes normalmente inactivos en
las células adultas. Por su parte, Huebner y Todaro propusieron en 1969 la hipótesis
del oncogene. Según esta hipótesis, las células de la mayoría de los vertebrados
contienen genomas correspondientes a virus ARN oncogénicos, incluyendo
las secuencias que codifican las actividades que pueden transformar a una célula
normal en célula tumoral (oncogenes). De acuerdo con esta hipótesis, las secuencias
correspondientes a los oncogenes son transmitidas en forma vertical de los progenitores
a la progenie y, por lo tanto, la aparición del cáncer será determinada por
la reactivación de esos oncogenes endógenos cuya expresión está normalmente
reprimida. Dicha reactivación puede ser inducida por carcinógenos químicos,
irradiación, envejecimiento celular o una combinación de todos estos factores.
Esta teoría ofrece una explicación para la frecuentemente observada transmisión
vertical de ciertos virus animales y la interacción cooperativa entre irradiación,
carcinógenos químicos, infecciones crónicas y los virus oncogénicos en la producción
de transformación celular.
Las dos teorías o hipótesis mencionadas proponen en común que el cáncer es consecuencia de la expresión de ciertos genes presentes en las células, mismos que normalmente no son expresados o lo hacen con muy baja intensidad; por lo tanto, el cáncer es consecuencia de una pérdida en la regulación de la expresión genética (figura IX.2.).
Los retrovirus han sido definidos como virus ARN que en forma
obligatoria se propagan a través de un intermediario intracelular constituido
por una molécula de ADN de cadena doble sintetizada por la enzima
transcriptasa inversa característica de estos virus, a partir del ARN
que constituye el genoma viral.
Si se supone que las secuencias correspondientes a los genes oncogénicos presentes
en los retrovirus tumorales no son necesarias para la replicación de estos virus,
como ocurre en el caso del RSV, sino que, por el contrario, constituyen
genes extras que proporcionan una cierta ventaja selectiva para la proliferación
celular, entonces se deduce que las células transformadas por retrovirus oncogénicos
deben contener secuencias de ácido nucleico que no están presentes en los retrovirus
no oncogénicos pertenecientes al mismo tipo o clase viral que los retrovirus
oncogénicos en cuestión. Esta idea predice que el genoma del virus transformante
(oncogénico) debe codificar cuando menos una proteína que está específicamente
asociada con el proceso de transformación y que esta proteína no es necesaria
para la replicación del virus a pesar de cumplir una función en las células
hospederas transformadas por el virus.
A principios de los años setenta diferentes grupos de investigadores encontraron
evidencia de que los virus del sarcoma aviario (de los cuales el RSV
es un ejemplo) contienen más información genética que la presente en cepas de
virus similares, pero que han perdido su capacidad para transformar células.
Las cepas de virus del sarcoma aviario (ASV) conocidas como "defectuosas
en transformación", no pueden inducir sarcomas en animales experimentales, tampoco
transforman células en cultivo y carecen de 10 a 20% de la información genética
(ARN) presente en virus similares, pero que cuentan con capacidad
transformante. Sin embargo, estos virus defectuosos en transformación son capaces
de infectar células y replicarse en forma normal dando origen a nueva progenie
viral. Esto indica que la eliminación de la porción del genoma que codifica
la actividad o actividades transformantes no tiene efecto alguno sobre la capacidad
del virus para replicarse. De hecho, la gran mayoría de los retrovirus directamente
oncogénicos, es decir, capaces de transformar directamente a la célula infectada
mediante la expresión de un oncogene viral, son virus "defectuosos en replicación".
O sea que estos virus oncogénicos no pueden replicarse porque han perdido funciones
virales esenciales para su replicación, ya que las secuencias génicas que codifican
dichas funciones han sido sustituidas por la secuencia correspondiente al oncogene
celular, mismo que fue adquirido por el genoma viral mediante un proceso mal
comprendido y que se conoce como "transducción". Por esta razón, la mayoría
de los retrovirus directamente oncogénicos sólo pueden ser propagados en presencia
de un virus auxiliar, el cual debe ser un retrovirus capaz de replicarse y,
por lo tanto, no es oncogénico. El retrovirus auxiliar aporta las funciones
virales que están ausentes en el retrovirus oncogénico, y así permite la replicación
y propagación de este último. Por lo tanto, la propagación de retrovirus directamente
oncogénicos es un fenómeno de laboratorio, ya que en condiciones naturales es
muy improbable que una misma célula sea coinfectada por el retrovirus oncogénico
y el retrovirus auxiliar. De hecho, no existen reportes de epidemias de cáncer
en animales silvestres.
 |
 |
 |
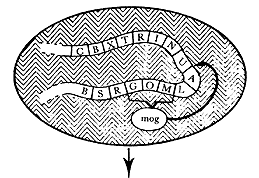
Figura IX. 2. Esquema que ilustra una posible manera de cómo el protovirus
puede causar una reorganización de los cromosomas de una célula para producir
información oncogénica. El protovirus transcribe un segmento del gene en ARN
(minúsculas), el cual es copiado en ADN e insertado en una nueva
posición dentro del cromosoma. La repetición de este proceso puede en algunos
casos producir un alineamiento paralelo de los genes (encasillados) que son
necesarios para producir carcinogénesis.
En 1976, Bishop, Varmus y colaboradores lograron aislar el fragmento del genoma
del virus del sarcoma de Rous (RSV) que codifica la actividad encargada
de inducir la transformación celular. Este gene fue bautizado como v-src.
El mismo grupo de investigadores demostró que en el genoma de células transformadas
por RSV y también en el genoma de células normales no infectadas
por éste virus, existen secuencias homólogas aunque no completamente idénticas
al v-src; estas secuencias homólogas fueron observadas en el genoma de
células de aves, ratones, peces, bovinos y humanos, pero no en células de erizo
de mar, mosca de la fruta ni en bacterias. Este resultado sugiere que una porción
del gene src ha sido conservada en el genoma de las células de organismos
superiores a lo largo de varias etapas de la evolución. Estudios posteriores
demostraron que ARN mensajero complementario a la secuencia presente
en el ADN correspondiente al gene v-src se encuentra presente
en el interior de células aviarias tumorales y normales. Esto indica la presencia
del gene src y la transcripción de este gene en ambos tipos de células.
Con el fin de evitar confusiones, el gene viral que codifica la función transformante
ha sido designado v-src, mientras que la secuencia de ADN
correspondiente a src y que está presente en el genoma de las células
normales ha sido designada como c-src. Otros experimentos demostraron
que la localización y concentración intracelular del ARNm correspondiente
al gene transformante v-src es de hecho la misma tanto en células transformadas
por RSV como en células que originalmente fueron transformadas
por RSV pero que han revertido en forma espontánea al estado normal.
Esto sugiere que la presencia del gene src y la transcripción del mismo
no son específicas de las células transformadas. Por lo tanto, si el gene src
está verdaderamente involucrado en el proceso de transformación, el factor esencial
debe estar localizado en el nivel de la traducción a proteína del ARNm
correspondiente al gene v-src o en el nivel del electo que tiene la proteína
codificada por v-src sobre ciertos componentes celulares.
La proteína codificada por el gene v-src ha sido identificada y se ha
encontrado que dicha proteína está presente tanto en células normales como en
células transformadas. Esta proteína fue aislada por métodos inmunológicos utilizando
el suero inmune obtenido de conejos que recibieron transplantes de tumores inducidos
por el virus del sarcoma aviario (ASV). Con este suero fue posible
precipitar una proteína con peso molecular de 60 000 daltones (p 60v-src),
a partir de extractos de células de pollo y de hámster que habían sido transformadas
por el ASV. Posteriormente, ha sido posible sintetizar la proteína
p 6-src in vitro a partir de
la región del ARN viral que contiene al gene v-src. La proteína
p 60v-src es
una fosfoproteína, o sea, tiene adosado un átomo de fósforo, el cual puede ser
despegado en forma reversible. Se dice que la proteína está fosforilada cuando
tiene el fósforo adherido a ella y esta forma de la proteína se denomina pp
60v-src. Curiosamente, la proteína p
60v-src tiene actividad de proteína cinasa,
o sea, es capaz de fosforilar a otras proteínas. La fosforilación ocurre en
los residuos del aminoácido tirosina presentes en la proteína a ser fosforilada.
Ésta es una característica muy peculiar, ya que otras proteína-cinasas conocidas
tienen los aminoácidos serina y treonina como blanco de la actividad fosforilante.
En células de pollo infectadas con el virus del sarcoma de Rous (RSV)
ha sido identificada una proteína con peso molecular de 36 000 daltones, la
cual en apariencia constituye el blanco (sustrato) de la actividad fosforilante
asociada con p 60v-src. Paradójicamente,
existe evidencia de que p 60v-src es
capaz de autofosforilarse. Por otra parte, se ha identificado en células normales
la presencia de una proteína fosforilada muy similar pero no idéntica a pp
60c-src. Dicha proteína es denominada
pp 60c-src, la concentración de esta proteína
en células normales se mantiene constante y no varía con el estado de crecimiento
celular; a diferencia de la proteína homóloga pp 60v-src,
la cual está presente en grandes cantidades en las células infectadas por el
ASV. Existe sólida evidencia de que la secuencia básica de aminoácidos
que constituyen la proteína pp 60c-src
ha sido conservada con mínimos cambios por diferentes especies a lo largo de
la evolución. El hecho de que en el gene c-src está presente la secuencia
de nucleótidos que codifica a la proteína p 60v-src
sugiere que el gene normal c-src presente en las células de varias especies
es el progenitor del gene v-src característico de los virus del sarcoma
aviario. Esta observación es consistente con la hipótesis del protovirus propuesta
por Temin. Hasta la fecha, no ha sido posible establecer con claridad la función
de p 6Oc-src en las células normales,
tampoco ha sido posible definir con claridad el papel que tiene p 60v-src
en la transformación celular. Es probable que esta proteína codificada por el
gene transformante presente en los virus del sarcoma aviario tenga otras funciones
aparte de su capacidad fosforilante, las cuales podrían estar más directamente
involucradas en el proceso de transformación celular.
En los últimos diez años diferentes grupos de investigadores han podido aislar otros genes virales con capacidad transformante a partir de diferentes tipos de retrovirus que afectan a diversas especies animales y producen diferentes tipos de tumores. En todos los casos estudiados hasta la fecha, ha sido posible encontrar en las células normales cuando menos un gene cuya secuencia de nucleótidos es muy similar a la del gene transformante presente en el genoma de cada tipo de retrovirus oncogénico estudiado. Estas observaciones apoyan en forma muy importante los postulados básicos de la hipótesis del oncogene celular propuesta por Huebner y Todaro.
Se deonomina proto-oncogenes a ciertos genes celulares que codifican diversas
funciones celulares muy importantes para el control de los procesos de diferenciación,
división y proliferación celular. Las diversas proteínas que son productos de
estos genes cumplen diversas funciones en las células: algunas actúan como factores
de crecimiento celular que son secretados al medio externo para que puedan estimular
a otras células; otras son receptores membranales capaces de ligar moléculas
que actúan como factores de crecimiento celular; otras proteínas transmembranales
que actúan como transmisores (transductores) de las señales generadas por la
interacción entre los factores membranales o citoplásmicas que pueden regular
la actividad de otras proteínas al fosforilarlas (cinasas de proteínas). Por
último, los productos de varios proto-oncogenes actúan directamente como reguladores
de la expresión de diversos genes, ya que son proteínas con capacidad para pegarse
a secuencias específicas del ADN celular, mismas que constituyen
las regiones promotoras o activadores de la expresión de ciertos genes.
Todos estos proto-oncogenes pueden convertirse en oncogenes cuando sufren mutaciones
que alteran su secuencia de nucleótidos. Las mutaciones pueden consistir en
la sustitución o eliminación de uno o varios nucleótidos, situación que modifica
la información codificada por el proto-oncogene y que resulta en la producción
de una proteína modificada y con función alterada. Sin embargo, también existen
las mutaciones causadas por inserciones génicas. Por ejemplo, el ciclo replicativo
de los retrovirus implica que el genoma viral, en forma de provirus de ADN,
debe integrarse en el genoma celular; dicha integración ocurre en sitios al
azar. Algunos retrovirus son indirectamente oncogénicos porque se integran cerca
de un proto-oncogene. Como consecuencia de esta integración puede ocurrir que
las secuencias promotoras de la expresión de los genes retrovirales, conocidas
como secuencias LTR, queden contiguas a la secuencia del proto-oncogene,
de manera que la interacción de estas secuencias promotoras con factores de
transcripción virales o celulares puede resultar en la hiperactivación del proto-oncogene,
el cual ahora se comporta como un oncogene que produce en forma anárquica el
ARN mensajero que codifica a una proteína normal, misma que al
encontrarse en exceso, induce la transformación celular. También puede ocurrir
que como consecuencia de un evento defectuoso de integración retroviral, un
segmento de un gene viral quede contiguo a un segmento de un proto-oncogene
celular, el resultado de este fenómeno es la creación de un gene quimérico,
mismo que codifica a una proteína quimérica la cual funciona en forma alterada
y puede desencadenar el proceso de transformación celular.
Fenómenos fisocoquímicos y ambientales como las radiaciones, pueden inducir rupturas y rearreglos en los cromosomas. Como resultado de estos rearreglos puede ocurrir que la secuencia de un proto-oncogene queda contigua a la secuencia promotora de la expresión de otro gene de mayor actividad. Esto resulta en la hiperactivación del proto-oncogene que se comporta como oncogene al ocasionar la sobreproducción de su proteína codificada, situación que provoca la pérdida de la regulación de la división y proliferación celular.
En 1978 se aisló por primera vez un retrovirus humano a partir de un cultivo de células conocidas como linfocitos T, procedentes de un paciente con un raro tipo de leucemia. Este retrovirus es ahora conocido como virus linfotrópico humano tipo 1 o HTLV-l, y es el prototipo de más de 100 diferentes muestras del mismo virus obtenidas alrededor del mundo. Antes, en 1977, Takatsuki y colaboradores habían descrito un raro tipo de cáncer conocido como leucemia de células T del adulto (ATL), el cual es relativamente frecuente en ciertas partes del sudoeste de Japón. El tipo y las características morfológicas de las células de este tumor son muy similares a las de las células a partir de las cuales se aisló el HTLV-1. Esta observación hizo sospechar que este virus podría ser el agente causal de la leucemia tipo ATL. En 1981, Gallo y colaboradores analizaron el suero de pacientes japoneses afectados por esta rara forma de leucemia y en el 100% de los casos encontraron la presencia de anticuerpos contra el HTLV-1. El siguiente paso consistió en el aislamiento del virus a partir de células obtenidas de pacientes con ATL. Esto fue logrado por Yoshida y colaboradores en 1982 y pronto fue demostrado que el virus de la leucemia ATL es idéntico al HTLV-1. Posteriormente fue posible identificar otras zonas y poblaciones del mundo en las cuales la presencia de este virus es endémica. En 1982 se descubrió un virus en particular prevalente en los monos verdes africanos; este virus, denominado virus linfotrópico de simios tipo 1 (STLV4), es homólogo en más de 95% al HTLV-l. Esta observación, asociada con datos referentes a la distribución geográfica del HTLV, ha hecho especular que el retrovirus humano es descendiente del retrovirus presente en los monos, el cual está asociado con la producción de linfomas malignos en los monos infectados. Estudios epidemiológicos, experimentos de transformación celular con linfocitos T normales y la caracterización de las propiedades moleculares del HTLV-1, sugieren que este virus puede estar involucrado en los mecanismos que causan la leucemia tipo ATL. Todas las células tumorales ATL contienen un genoma de HTLV-1 en forma de provirus, mismo que no está presente en las células normales. El provirus es clonal, o sea, es exactamente igual en todas las células tumorales; esto indica que cada provirus es descendiente directo del provirus progenitor presente en la célula que dio origen al tumor. Este hecho implica que la infección por HTLV-1 ocurre antes del primer evento de transformación celular, mismo que dará origen a la primera célula tumoral. El sitio de integración del provirus dentro del genoma celular es el mismo en todas las células, pero varía de un paciente a otro ya que en diferentes enfermos el tumor se origina a partir de un linfocito T diferente. La figura IX.3 ilustra tres diferentes modelos propuestos para explicar la producción de leucemia por tres diferentes tipos de retrovirus.
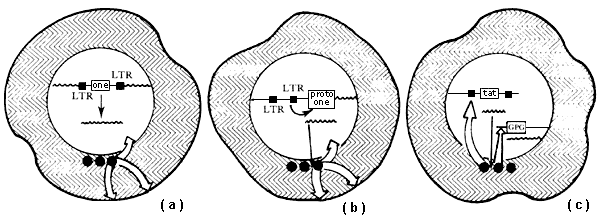
Figura IX.3. Mecanismos de leukemogénesis viral. (a) Los virus productores
de leucemias agudas capturan genes derivados de las células (oncogenes) que
codifican proteínas específicas involucradas en la transformación celular. Algunas
de estas proteínas son promotoras del crecimiento celular y pueden estar localizadas
en la membrana, el citoplasma o el núcleo celular. Estos posibles sitios de
acción son indicados por las flechas. Estas proteínas actúan en trans,o
sea, sobre una región del ADN diferente a la que contiene el oncogene
y el provirus integrado; por lo tanto, no se requiere de un sitio constante
y específico para la integración del provirus. (b) Los virus productores de
leucemias crónicas activan genes celulares a causa de que se integran en forma
de provirus en una posición próxima a los genes afectados. Los genes celulares
activados pueden ser proto-oncogenes. El mecanismo de activación en cisrequiere
de la conservación de sitios de integración proviral específicos. (c) El virus
HTLV-1, asociado con la leucemia tipo T del adulto (ATL), produce una proteína
nuclear (TAT) que activa la transcripción de genes localizados en segmentos
del genoma diferentes a los que contiene el provirus integrado (activación en
trans);por lo tanto, no es necesario que exista un sitio específico para
la integración del provirus. Se ha especulado que el producto del gene ta
tes capaz de activar la expresión de genes promotores del crecimiento celular
(GPG) específicos para células linfoides. Las letras LTR indican las secuencias
repetidas invertidas que están presentes en ambos extremos del genoma correspondiente
al provirus integrado o al virus HTLV-1 no integrado.
Desde la época grecoromana se sospechaba que las verrugas en la región anogenital
del humano podían ser de origen venéreo. El origen infeccioso de estas verrugas
fue establecido a fines del siglo XIX, y en 1907 Ciuffo demostró
la transmisión de los papilomas epidérmicos, por medio de la inoculación de
voluntarios con filtrados libres de células obtenidas a partir de estos papilomas
(las verrugas o tumores benignos conocidos como "mezquinos"). Esto demostró
la etiología viral de los papilomas (mezquinos). En 1932, Shope aisló el virus
del papiloma de conejo, mismo que produce papilomas y tumores en la piel del
conejo. Este virus fue el primer modelo para estudiar el posible papel oncogénico
de los virus en los mamíferos. Desde entonces empezó a sospecharse que puede
existir una cooperación entre ciertos virus y carcinógenos químicos durante
el desarrollo de ciertos tipos de tumores.
El estudio de los virus del papiloma humano (VPH) o Human Papilloma Virus (HPV) ha estado limitado por la falta de sistemas para cultivarlo in vitro, ya que este tipo de virus solamente infecta células epiteliales y sólo se replica activamente en células epidérmicas (queratinocitos) totalmente diferenciadas, mismas que no pueden ser mantenidas en cultivo por largo tiempo. Sin embargo, el advenimiento de las técnicas de ingeniería genética ha permitido "donar" los genomas de varios tipos de virus del papiloma humano HPV, obtenidos a partir de biopsias de tejidos infectados. Hasta la fecha han sido identificados 68 genotipos diferentes de HPV. Por convención, se considera que dos cepas de HPV constituyen tipos diferentes si la secuencia de nucleótidos de sus genomas manifiestan una homología menor a 50%.
Existen virus del papiloma en diferentes especies animales y cada uno de estos
virus es específico para la especie a partir de la cual fue aislado, o sea que
el HPV no infecta especies animales y los virus de animales no pueden infectar
al humano. Todos los virus del papiloma (papilomavirus) pertenecen al grupo
de los papovavirus y se caracterizan por ser virus sin envoltura con cápside
icosaédrica y con un genoma circular de ADN de doble cadena que
consta de aproximadamente 8 000 pares de bases. Las infecciones por HPV tienen
una dis tribución mundial.
Se ha podido establecer que la infección por HPV tipo 5 está muy asociada con
el desarrollo de cáncer de piel en pacientes que padecen una rara enfermedad
congénita llamada epidermodisplasia verruciforme (EV). Por otra parte, desde
mediados del siglo XIX se ha sospechado que el cáncer del cérvix
(cuello) uterino puede ser de origen infeccioso. Estudios epidemiológicos realizados
entre 1960 y 1970 parecían implicar al virus Herpes simplex tipo 2 en
la etiología de este tipo de cáncer; sin embargo, en 1974 surgió la primera
evidencia de una asociación entre la infección por HPV y el subsecuente desarrollo
de cáncer cérvico uterino (CaCu). En 1984 el grupo dirigido por Harald zur Hausen
reportó el aislamiento frecuente de los HPV 16 y 18 a partir de tumores cervicouterinos.
En la actualidad se sabe que el HPV 16 está presente aproximadamente en de 50%
de los CaCus, mientras que el HPV 18 se encuentra cerca del 20% de estos tumores.
Otros tipos de HPV han sido encontrados en muestras de CaCu, por lo cual casi
el 90% de los tumores cervicouterinos son positivos a HPV. El periodo de latencia
entre la infección primaria y la aparición del cáncer es del orden de 20 a 40
años. Esta situación es similar a la observada en otros tumores humanos asociados
con infecciones virales como son el cáncer de hígado con el HBV y la leucemia
de células T del adulto con el HTLV-1.
En la mayoría de las lesiones pretumorales asociadas con infección por HPV,
se observa que el ADN viral persiste en forma de episoma o sea,
como un minicromosoma independiente del resto del genoma de la célula hospedera.
Sin embargo, en la mayoría de los tumores cervicouterinos se observa la integración
del genoma viral, este evento de integración parece ser uno de los eventos tempranos
en el proceso de carcinogénesis. El patrón de integración con frecuencia resulta
en la interrupción de la secuencia correspondiente al gene E2 del HPV. La proteína
codificada por este gene está involucrada en la regulación de la expresión de
otros genes del HPV. La ausencia de la proteína E2 facilita la sobreexpresión
de los genes virales E6 y E7. Se sabe que las proteínas codificadas por E6 y
E7 son capaces de interactuar con las proteínas celulares p53 y RB respectivamente,
dichas proteínas celulares participan en los mecanismos que controlan la división
y proliferación celular, la inactivación de estas proteínas celulares está asociada
con el desarrollo de varios tipos de tumores; Por lo anterior; se piensa que
el mecanismo oncogénico del HPV puede estar relacionado con la inactivación
de p53 y RB por medio de ciertas proteínas virales.
En los últimos años se han incrementado los reportes que asocian la infección por HPV en epitelios extragenitales con el subsecuente desarrollo de tumores de la laringe, amígdalas, lengua y cavidad oral. Lo anterior sugiere que el potencial oncogénico de estos virus puede afectar diversos órganos humanos. La frecuente regresión espontánea de papilomas y verrugas benignas, sugiere que el sistema inmune puede controlar en la mayoría de los casos la infección por HPV. Sin embargo, se sabe muy poco respecto a la respuesta inmune específica contra el HPV, ya que esta respuesta inmune no parece ser intensa debido a que los HPV provocan infecciones crónicas y latentes, y en el caso de las infecciones productivas éstas no se asocian con la destrucción de la célula infectada, ya que los HPV no son citopáticos. Es probable que la inmunidad mediada por células (inmunidad celular) sea la que tenga un papel más importante en el control de las infecciones por HPV. Lo anterior se deduce de la alta incidencia de papilomas, verrugas y tumores epiteliales observada en pacientes que sufren de inmunodeficiencias congénitas o adquiridas. Así, una prioridad de la investigación sobre los HPV consiste en lograr un método para estimular la inmunidad celular específica contra los HPV.
La hepatitis es un padecimiento relativamente frecuente que se manifiesta como una inflamación generalizada del hígado con la consecuente alteración de importantes funciones metabólicas que tienen lugar en las células hepáticas (hepatocitos). La hepatitis humana de origen viral puede ser causada por una importante variedad de virus; sin embargo, existe un grupo de virus que manifiesta una gran predilección por infectar el tejido hepático (tropismo hepático). Hasta hace poco tiempo, las hepatitis humanas causadas por virus con tropismo hepático se clasificaban en tres tipos: A, B y no A-no B. Esto se debía a que solamente habían sido identificados los virus de la hepatitis tipo A (HAV) y de la hepatitis B (HBV). Hoy sabemos que las hepatitis virales no A-no B pueden ser causadas por, cuando menos, tres virus diferentes: HCV, HDV y HEV.
Podemos dividir a las hepatitis virales específicas en dos grupos principales: 1) hepatitis virales de incubación corta, las cuales se adquieren por lo general a través del contagio oral-fecal (principalmente por ingestión de agua contaminada por heces o de alimentos contaminados por dichas aguas residuales) y que se comportan como hepatitis agudas de resolución rápida. Los virus HAV y HEV son los principales causantes de estas hepatitis. 2) hepatitis virales de incubación larga, las cuales se adquieren por lo general a través de contagio por vía parenteral: por transfusiones con sangre contaminada, por contagio sexual, por heridas causadas por instrumentos quirúrgicos contaminados y en el caso de los toxicomanos, por compartir jeringas y agujas contaminadas. También se ha reportado la transmisión de la madre al hijo durante el periodo perinatal. Estas hepatitis evolucionan con frecuencia hacia formas de infección crónica y persistente, y los pacientes afectados pueden sufrir recaídas repetidas, además de continuar siendo contagiosos durante muchos años. Las hepatitis virales de origen parenteral e incubación larga son causadas por los virus HBV, HCV y HDV.
Es interesante notar que los cinco virus de la hepatitis pertenencen a cinco grupos taxonómicos diferentes, o sea que desde el punto de vista de su organización estructural, de la organización de sus genomas y de sus respectivas estrategias de replicación, estos cinco virus son muy diferentes entre sí y solamente comparten la predilección por infectar el hígado. El virus HAV pertenece al grupo de los picornavirus cuyo miembro más conocido es el virus de la poliomelitis. El virus HCV pertenece al grupo de los flavivirus cuyos miembros más conocidos son causantes de la fiebre hemorrágica conocida como "dengue". Se sabe que varios tipos de flavivirus pueden ser transmitidos por insectos (artrópodos), sin embargo, no existe ninguna evidencia de que el HCV pueda ser transmitido por insectos. El HEV pertenece al grupo de los calcivirus, entre los cuales se encuentran virus que afectan a los felinos y otros virus com el Norwalk que causa enfermedades diarréicas en los humanos.
El espacio de la presente obra no permite una discusión detallada de los cinco virus de la hepatitis; sin embargo, debido a su importancia médica y a las interesantes peculiaridades de sus modos de organización y replicación, vale la pena dedicar algunos párrafos a los virus HBV y HDV.
En 1963, el investigador Baruj Blumberg descubrió en el suero sanguíneo de
un paciente con hemofilia (padecimiento cuyo tratamiento requiere de repetidas
transfusiones de sangre o plasma sanguíneo), la presencia de un anticuerpo que
era capaz de reaccionar con un antígeno presente en la sangre de un aborigen
australiano. Blumberg denominó antígeno Australia a esta molécula que resultó
ser el antígeno principal de superficie del virus de la hepatitis B (HBsAg).
La ausencia de cultivos celulares específicos capaces de replicar al HBV
in vitro, impidió la pronta caracterización de este virus. Sin embargo,
con el advenimiento de las técnicas modernas de ingeniería genética, fue posible
aislar y "domar" el genoma de HBV con el fin de estudiar la biología molecular
de este virus. A finales de la década de los setenta, investigadores franceses
pudieron establecer que el genoma del HBV consta de un ADN circular
de doble cadena (aunque es desigual la longitud de ambas cadenas), correspondiente
a 3 200 pares de bases, siendo hasta la fecha el genoma más pequeño de todos
los descritos en virus animales.
El HBV resultó ser el primer tipo descrito de un nuevo grupo de virus denominado
hepadnavirus, debido a que todos los virus que son miembros de este grupo manifiestan
un marcado tropismo hepático y comparten un método de replicación muy particular,
mismo que es descrito en la figura IX. 4. Entre los virus que pertenecen a este
grupo se encuentran virus que causan hepatitis en marmotas, ardillas y patos.
Cabe mencionar que los hepadnavirus son los únicos virus, aparte de los retrovirus,
que incluyen en su ciclo de replicación la actividad de una enzima transcriptasa
inversa, capaz de sintetizar una molécula de ADN a partir de un
templado de ARN. De hecho, la polimerasa codificada por el gene
P de los hepadnavirus, es una enzima que manifiesta cuatro actividades diferentes:
ADN polimerasa dependiente de ADN; ADN
polimerasa dependiente de ARN (transcriptasa inversa); Ribonucleasa
H, capaz de degradar el ARN presente en moléculas híbridas ADN-ARN;
actividad como molécula anda para la iniciación de la síntesis de ADN.
Otra característica importante de los hepadnavirus consiste en que no son virus directamente citopáticos, o sea que no destruyen a su célula hospedera. El conocimiento actual sobre la hepatitis tipo B indica que el daño hepático, que se manifiesta como inflamación hepática y destrucción de los hepatocitos, es causado por la propia respuesta inmune dirigida contra las células infectadas por el HBV; en particular, los linfocitos T citotóxicos anti-HBV parecen ser los principales responsables de la destrucción de los hepatocitos que expresan antígenos (proteínas) de HBV en sus membranas. Se piensa que los casos de hepatitis B fulminante y fatal, son consecuencia de una excesiva respuesta inmune contra el HBV, aunque en ratones transgénicos, en cuyas células se les ha insertado en forma experimental el gene que codifica el HBsAg, se observa que la sobreproducción del HBsAg puede causar destrucción de los hepatocitos.
Sin embargo, la mayoría de las personas infectadas por HBV desarrollan un cuadro de hepatitis aguda, después de un periodo de incubación de ocho a 24 semanas. Dicha hepatitis aguda se manifiesta con dolor abdominal, ictericia (coloración amarillenta de la piel), fatiga y otros síntomas asociados. En otros casos, también numerosos, la infección permanece asintomática; pero en todos los pacientes en fase aguda de la infección (sintomática o asintomática) es posible detectar la presencia de altas concentraciones del HBsAg en el suero sanguíneo. Con el paso del tiempo y en la medida en que disminuye la hepatitis, aparecen anticuerpos específicos contra HBsAg (anti-HBs) en el suero de los pacientes infectados.
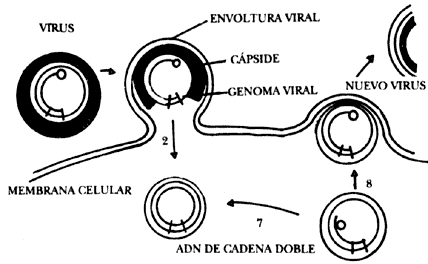
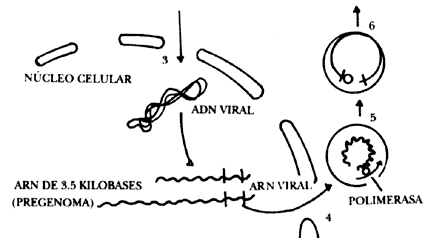
Figura IX. 4. Replicación del virus de la hepatitis B (HBV)1)
El HBV infecta a una célula hepática. 2) Enzimas extienden la cadena corta del
ADN viral. 3) El ADN viral migra hacia el núcleo celular, donde
es copiado (transcrito) en una molécula complementaria de ARN.
La hebra de ARN tiene 3.5 kilobases (3 500 nucleótidos) y constituye
el pregenoma que es intermediario necesario para la replicación del genoma viral.
4) El pregenoma es empacado dentro de una cápside recién sintetizada. La polimerasa
viral empieza a sintetizar una copia de ADN complementario al pregenoma
viral. 5) La nueva cadena de ADN es un duplicado de la cadena larga
de ADN presente en el genoma viral original. El pregenoma de ARN
se desintegra tan pronto es completada la síntesis del nuevo ADN
viral. 6) La polimerasa viral empieza a sintetizar una cadena de ADN
complementario a la secuencia de nucleótidos de la cadena larga del ADN
viral. 7) El ADN viral puede permanecer en la célula por un tiempo
suficiente para convertirse en ADN de cadena doble; en cuyo caso,
regresa al núcleo celular para iniciar un nuevo ciclo de replicación. 8) En
caso de salida prematura de la nueva partícula viral, la cápside es dotada de
una nueva envoltura y la extensión de la cadena corta del ADN viral
cesa tan pronto la nueva partícula sale de la célula infectada. El resultado
es una partícula viral infectante que contiene una ADN viral que
es parcialmente de cadena sencilla.
Por lo general, estos anticuerpos alcanzan la concentración suficiente para neutralizar las partículas infecciosas de HBV y se sostienen a buenos niveles circulantes, de manera que protejen de por vida contra una nueva infección por HBV. Sin embargo, en algunos pacientes la respuesta inmune es deficiente y esto permite que persistan altas concentraciones de HBsAg circulante en el suero de estos pacientes. El propio HBV persiste en el hígado de tales pacientes, mismos que se convierten en portadores crónicos de este virus y tienden a presentar cuadros recurrentes de hepatitis (recidivas) tanto sintomática como asintomática. La sangre de estos portadores crónicos es una fuente potencial de contagio para los individuos que no están infectados por HBV.
Cabe subrayar que los hepatocitos infectados por HBV producen viriones infectantes completos (partículas de Dane), pero también producen partículas virales defectuosas, tanto esféricas como cilíndricas, que carecen del genoma viral pero que presentan en su superficie el antígeno principal del HBV (HBsAg), en sus tres versiones: grande, mediana y corta o principal. De hecho, en toda infección por HBV la producción de partículas defectuosas supera a la de viriones completos por varios órdenes de magnitud. Lo anterior indica en forma indirecta que los viriones completos tienen una gran capacidad infectiva y son suficientes para propagar la infección.
Un aspecto muy importante, desde el punto de vista médico, es que alrededor del 30% de los portadores crónicos de HBsAg desarrolla cirrosis hepática, la cual es una degeneración del tejido hepático que es sustituido por tejido cicatrizal que no es funcional. De estos pacientes con cirrosis, alrededor del 30% desarrolla un cáncer primario de hígado, evento que puede ocurrir entre 30 y 50 años después de la infección original por HBV. Estudios epidemiológicos demuestran que las posibilidades de convertirse en portador crónico se incrementan entre más temprana es la edad en que se contrae la infección por HBV. Se considera que la probabilidad de que un portador crónico de HBsAg desarrolle un cáncer de hígado es 200 a 300 veces mayor que en el resto de la población. Por esta razón, la infección temprana por HBV constituye uno de los problemas mas importantes de salud pública mundial, ya que en los países del Centro y sur de África y en los países del Lejano Oriente, la infección por HBV ocurre con una gran frecuencia en niños recién nacidos (que son infectados por sus madres durante el embarazo o durante el periodo perinatal) y en menores de cinco años. Se estima que en el mundo viven actualmente 300 millones de portadores crónicos del HBsAg, los cuales son también portadores crónicos del HBV, esto significa que a partir de esta población infectada se van a desarrollar cerca de 30 millones de casos de cáncer hepático. De modo que el HBV es en términos estadísticos el agente biológico con mayor potencial oncogénico (productor de cáncer) para los seres humanos.
El largo periodo de latencia entre la infección original por HBV y la aparición
del cáncer hepático, no es compatible con la hipótesis de que este cáncer es
causado por la activación de un oncogene presente en el genoma de HBV. De hecho,
los estudios de biología molecular demuestran que el virus de la hepatitis B
no posee ningún oncogene; además, está bien establecido que el HBV no necesita
integrar su ADN en el genoma de la célula hospedera para llevar
a cabo su ciclo de replicación. Por otra parte, diversos estudios han reportado
la presencia de fragmentos de genoma del HBV integrados en el ADN
de células procedentes de tumores hepáticos. El análisis de estas células sugiere
que los fragmentos del genoma viral pueden integrarse en sitios diversos del
genoma celular y de esta manera pueden provocar rearreglos de secuencias de
nucleótidos o modificar el patrón de expresión de ciertos genes celulares. También
se ha reportado que la sobreproducción de la proteína HBx codificada por el
gene X del HBV puede inducir tumores de hígado en ratones que han sido genéticamente
modificados para albergar y expresar el gene X dentro de sus células (ratones
transgénicos). Se sabe que la proteína HBx puede actuar como activadora de la
expresión de genes virales y celulares.
Sin embargo, la mayoría de los estudios epidemiológicos sugiere que el desarrollo del cáncer hepático requiere de un evento genético secundario que es consecuencia de los ciclos de destrucción y regeneración hepática característicos de la hepatitis crónica y la cirrosis. Esto pone en duda que el cáncer hepático sea consecuencia directa de la acción de un gene viral. En realidad todo parece indicar que los tumores hepáticos asociados con la infección crónica por HBV son causados por una combinación de factores: daño hepático, regeneración celular y actividades virales que actúan en forma sinergista para producir inestabilidad cromosómica con el paso del tiempo, inestabilidad que se manifiesta como una modificación gradual de las funciones y el estado de diferenciación de los hepatocitos. El hecho bien demostrado de que agentes hepatotóxicos como el alcohol y las aflatoxinas, producidas por ciertos hongos que contaminan a cereales y semillas, aceleran el desarrollo de la cirrosis hepática y la progresión hacia el cáncer hepático en individuos crónicamente infectados por HBV, apoya la idea de que el virus necesita de cofactores que actúan por periodos prolongados para producir el cáncer hepático.
Se han utilizado algunos agentes antivirales como tratamiento para los cuadros de hepatitis crónica recurrente asociada con la infección por HBV. Sin embargo, solamente el interferón alfa, producido por técnicas de ingeniería genética, ha demostrado tener un efecto positivo, aunque no es curativo. Afortunadamente, desde hace algunos años existen vacunas eficaces contra el HBV. La primera de ellas, desarrollada en Francia a finales de los años setenta, se basa en la purificación de envolturas y partículas virales defectuosas a partir del suero sanguíneo de portadores crónicos del antígeno HBsAg. Dichas partículas no son infectantes; sin embargo, poseen el antígeno mayor de superficie del virus (el HBsAg), por lo cual la inyección de estas partículas purificadas provoca la producción de anticuerpos neutralizantes específicos contra HBsAg en los individuos vacunados. En la actualidad, también están disponibles vacunas recombinantes o sea, vacunas producidas por métodos de ingeniería genética. Para desarrollar estas vacunas se procedió a donar el gene del HBsAg en diferentes tipos de vectores de expresión, mismos que permiten introducir dicho gene a células de mamífero o levaduras que son utilizadas como "fábricas" para la producción de dicho antígeno, el cual es purificado en forma subsecuente y utilizado para preparar vacunas que inducen anticuerpos específicos contra HBsAg.
Desafortunadamente, el alto costo de estas vacunas hace que su disponibilidad sea muy limitada, ya que se utilizan principalmente en los países desarrollados y en programas de vacunación destinados a proteger personal de alto riesgo como son los soldados, los médicos y paramédicos. Por otra parte, los niños africanos y asiáticos que corren gran riesgo de ser infectados por HBV durante los primeros años de vida constituyen, por lo tanto, el principal reservorio de este virus y el principal grupo que será afectado por la cirrosis y el cáncer de hígado; permanecen, hasta el momento, fuera de cualquier programa mundial de vacunación dirigido a controlar la infección por RBV.
Agente delta o virus de la hepatitis delta
El agente etiológico de la hepatitis delta, mal llamado virus de la hepatitis delta o HDV, fue descubierto por Rizzeto en 1977, pero su caracterización fue posible hasta 1986 gracias a la biología molecular. Este agente es capaz de infectar los hepatocitos exclusivamente en presencia y con la ayuda del virus de la hepatitis B; de tal manera que los individuos inmunizados contra el virus B, ya sea por vacunación o por inmunidad adquirida después de una infección por HBV, quedan protegidos también contra el HDV.
El genoma del agente delta consta de 1678 nucleótidos que forman una cadena
de ARN circular. Las características moleculares del HDV se parecen
mucho a las de los viroides que son moléculas de ARN desprovistas
de cápside y que, sin embargo, son capaces de producir diversas enfermedades
en las plantas. Hasta el momento, todo parece indicar que el genoma del agente
delta sólo codifica una proteína conocida como antígeno delta (HDAg), la cual
tiene una gran afinidad por el propio ARN de este viroide y también
por el antígeno principal del HBV (HBsAg). Estas propiedades del HDAg permiten
que el genoma del viroide sea empacado dentro de envolturas compuestas por lípidos
de la célula infectada y múltiples copias del HBsAg; es decir, este viroide
utiliza al antígeno principal del HBV para conformar su propia envoltura y así
poder propagarse como un agente infeccioso. Por lo anterior, la infección por
el agente delta sólo puede prosperar en individuos que han sido coinfectados
con HBV y HDV al mismo tiempo, o que padecen de hepatitis B crónica o son portadores
crónicos del HBsAg. En todos estos casos, el HBV puede actuar como virus auxiliar
al proporcionar el material necesario (HBsAg) para el empacamiento de nuevas
partículas infecciosas del agente delta.
La infección por HDV agrava los cuadros de hepatitis crónica en pacientes portadores de HBV y se han reportado casos de hepatitis fulminante en portadores crónicos del HBV que han sido superinfectados por el HDV. Por lo general, los individuos infectados con HDV presentan anticuerpos contra el antígeno delta (HDAg) durante un periodo muy corto después de la infección. Sin embargo, los portadores crónicos del HBV que son superinfectados por el HDV evolucionan con frecuencia hacia una hepatitis crónica que se acompaña de la continua presencia del HDAg en el hígado y de anticuerpos anti-HDV en la sangre. La mayor parte de estas hepatitis crónicas degenera en cirrosis hepática. Las partículas de HDV presentan en su superficie al antígeno HBsAg; por esta razón, los pacientes infectados por HDV desarrollan anticuerpos contra HBsAg, mismos que son indistinguibles de aquellos desarrollados por pacientes infectados únicamente por HBV. Estudios realizados en chimpancés (que son los únicos animales aparte del hombre susceptibles a la coinfección por HBV y HDV), sugieren que la vacunación con preparaciones a base de HDAg no protege de la infección por el agente delta y por el contrario, parece agravar el curso de esta infección.