II. CÁNCER Y HERENCIA
¿CÓMO SE REGULAN LAS SEÑALES MODULADORAS DE LA PROLIFERACIÓN Y DIFERENCIACIÓN CELULAR?
Uno de los grandes secretos de la vida es la forma en la que se ha generado, conservado, diversificado y transmitido la información que permite el surgimiento de los seres vivos —y su extraordinaria diversidad—, y que asegura además la constancia de las características que definen a cada especie en particular (o sea, cómo se ha estructurado y funciona la información hereditaria).
Es ahora de todos conocido que los seres vivos, cualquiera que sea su naturaleza, poseen información hereditaria contenida en los ácidos nucleicos, los cuales constituyen el material genético. Estos ácidos nucleicos son el ácido desoxirribonucleico (ADN), el de la mayoría de los seres vivos, y el ácido ribonucleico (ARN), el de ciertos virus. Los componentes fundamentales de dichos ácidos son los nucleótidos, constituidos por cuatro bases nitrogenadas: adenina, guanina, citosina y timina (que en el ARN es remplazada por uracilo), más un azúcar (desoxirribosa en el ADN y ribosa en el ARN) y un ácido fosfórico. Dichos nucleótidos son como las letras del alfabeto con el que se construye el código genético donde se almacena la información hereditaria.
También nos es familiar que el ADN adopta la forma de dos cadenas que se enrollan en forma similar a la de una escalera de caracol, en la que los peldaños están representados por el apareamiento de dos bases nitrogenadas (siempre adenina frente a timina, y guanina frente a citosina). El ADN de las células humanas se encuentra superenrollado, compactado y repartido en 23 pares de estructuras que conocemos como cromosomas y contiene segmentos con combinaciones específicas de las cuatro bases, las cuales constituyen unidades de información conocidas como genes (Figura 5). Se sospecha que poseemos alrededor de 100 000 genes en lo que llamamos el genoma humano; dichos genes controlan la síntesis de otras tantas proteínas (o de las cadenas de aminoácidos de los polipéptidos que las forman), las cuales caracterizan a las células y desempeñan un papel importante en su estructura y funcionamiento.
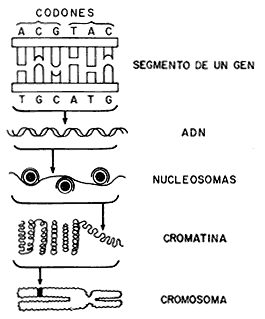
Figura 5. Organización del material genético.
En la regulación de la lectura del mensaje hereditario contenido en los genes parece estar encerrada la clave del secreto de la vida al que hacíamos referencia. Ésta podría ser una forma de interpretar la observación de que nunca en la vida de una célula se leen simultáneamente todos sus genes, sino que, por el contrario, su lectura es dosificada de tal modo que permita esa maravillosa diversidad de formas y funciones de las células que nos conforman. La regulación de la información hereditaria se parece a la elaboración y lectura de un texto en donde es importante tanto el orden y combinación de las palabras como las pausas y silencios al leer las frases y obedecer las puntuaciones.
Esta misma forma de regulación se aplica a los procesos que llevan a la célula a dividirse y diferenciarse, y en los que las moléculas proteicas clave que participan en ellos también están sujetas a los mecanismos de control que determinan cuándo y cuánto deben leerse los genes que contienen la información para su síntesis. Hoy en día sabemos que diversas moléculas proteicas a las que se hace referencia están codificadas por los llamados protooncogenes, o precursores de los genes del cáncer u oncogenes.
|
Los oncogenes son versiones alteradas de genes que
participan en las funciones de regulación de la proliferación y diferenciación
celular (protooncogenes) y que se expresan de manera inadecuada en momentos
inoportunos o en células no apropiadas.
|
Es importante mencionar que, aunada a la regulación genética de las señales que participan en la modulación de la división y diferenciación celular, existe otra forma de regulación que se desencadena cuando el factor de crecimiento se une a su receptor. Se trata de los procesos de fosforilación que se repiten una y otra vez a distintos niveles para lograr, mediante la introducción de grupos fosfato en los aminoácidos clave de las proteínas, ya sea su activación o, por el contrario, el bloqueo de sus funciones. De esta manera, el factor de crecimiento produce un pulso que da lugar a una sola división celular, ya que las proteínas activadas directa o indirectamente (incluyendo la que constituye su propio receptor), al final del proceso se inactivan mediante fosforilación de un aminoácido específico (tirosina, figura 6), con lo que se apaga el circuito. Esto hace que una célula normal tenga que esperar la llegada de una nueva señal para volver a dividirse. En las células cancerosas esta forma de regulación está también alterada, pues algunas de las moléculas proteicas que deberían ser inactivadas han perdido, en el sitio preciso, el aminoácido clave que requiere ser fosforilado para apagarlas.
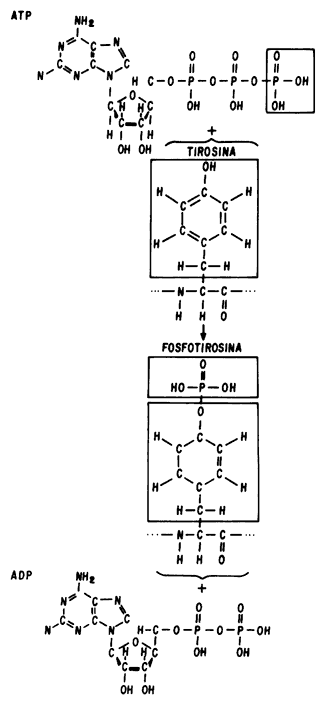
Figura 6. Fosforilación de tirosina.
Todo lo anterior muestra la versatilidad de la fosforilación, ya que la introducción de grupos fosfato constituye un interruptor que, según donde ocurra, provoca el encendido o el apagado del proceso de división celular.
El proceso de fosforilación también permite entender por qué una pequeña mutación en el material genético, la cual da lugar a un cambio de un aminoácido por otro en una proteína participante en la división celular, puede hacer que las células proliferen continuamente al perderse el sitio en el que se introduce un grupo fosfato para bloquear su actividad.
|
El proceso que desencadena la división celular es
como un bello fuego de artificio que se enciende con una flama y poco
a poco, una tras otra, va alumbrando figuras y culmina con la aparición
de una imagen para luego, en un instante, apagarse por completo. Por
el contrario, en las células cancerosas el sistema permanece encedido
todo el tiempo.
|
¿CÓMO SE DESCUBRIERON LOS ONCOGENES?
La historia del descubrimiento de los oncogenes está ligada con la de la identificación del agente viral que causa el llamado sarcoma de Rous en las aves. Hace cerca de 80 años el patólogo Francis Peyton Rous decidió inyectar a docenas de gallinas con el filtrado que obtuvo al tamizar, a través de una malla muy fina, una suspensión de células de un tumor obtenido por autopsia de una gallina de la misma raza. Con este procedimiento logró reproducir el tumor y sospechó que el agente causal debería ser de menor tamaño que las células y que las bacterias, por lo que podría corresponder a un virus, aunque no lo designó así sino como un agente tumoral. Cabe mencionar que el doctor Rous recibió el premio Nobel de Medicina por sus descubrimientos en 1966.
Más recientemente el doctor J. Michael Bishop, galardonado en 1989 también con el Premio Nobel de Medicina, informó que el gen v-src, el cual permite al virus causante del sarcoma de Rous inducir el tumor, también está contenido en las células normales no tan sólo de la gallina sino de algunos vertebrados, incluyendo al ser humano. Logró este descubrimiento gracias al empleo de las nuevas tecnologías de la llamada ingeniería genética, que permitieron, en primer lugar, copiar al gen v-src mediante una enzima que puede generar una versión de ADN a partir de la hebra de ARN del genoma viral (transcriptasa reversa). El siguiente paso consistió en multiplicar el número de copias del gen v-src, marcándolas con un isótopo radioactivo para poder seguir su destino. Finalmente las puso en contacto con el ADN desnaturalizado (separado en sus dos hebras) extraído de los distintos tipos celulares. Esto trajo como resultado la formación de algunas cadenas híbridas (que contenían una hebra de ADN marcado, la copiada del genoma viral, y otra sin marcar, proveniente de la célula donadora), que revelaron zonas de homología y descubrieron la misma secuencia en las células normales.
El estudio de la secuencia de nucleótidos del gen src permitió, además, descubrir que, por su organización (presencia de secuencias conocidas como exones e intrones típicas de los genes animales pero no virales), ese gen no pertenecía al virus, sino que debió ser arrastrado por éste después de unirse y desprenderse del material genético de alguna célula hospedera (Figura 7). Más aún, sus homólogos en las células normales resultaron ser genes activos y fue también el grupo del doctor Bishop quien descubrió la proteína codificada por el gen src (llamado c-src porque corresponde a la versión celular de ese gen), a la que denominaron pp60c-src, y que, sorprendentemente, resultó ser una proteína fuertemente unida a la superficie interior de la membrana celular y capaz de fosforilar a las tirosinas.
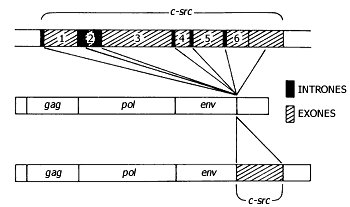
Figura 7. Incorporación del oncogén src al genoma viral.
Hoy se sabe que en las células cancerosas que contienen activo el oncogén c-src: está presente una proteína pp60c-src a la que, curiosamente, le falta, en un sitio peculiar, una tirosina, que en su versión normal es fosforilada, como mecanismo para bloquear la propia actividad fosforilante de la proteína pp60c-src. Tal parece ser la explicación por la cual esa proteína está permanentemente funcionando e introduciendo grupos fosfato en otras proteínas, en el caso de las células tumorales.
|
Ciertos virus pueden secuestrar porciones del material
genético de las células a las que infectan llevándolo consigo de una
célula a otra como si fuera propio.
|
¿QUÉ SE HA APRENDIDO DE LOS VIRUS QUE PRODUCEN CÁNCER?
Los virus cuyo material genético está constituido por ARN y que tienen la capacidad de inducir la síntesis de la enzima transcriptasa reversa, pueden hacer copias de ADN a partir de su ARN e insertarse en el genoma de las células hospederas como lo hace el virus del sarcoma de Rous. A estos virus se les conoce como retrovirus y constituyen una familia a la cual pertenece el virus del sida y otros que no causan enfermedades y que parecen formar parte integrante de algunas células y participar en la regulación de sus procesos de diferenciación.
Los retrovirus que producen cáncer lo hacen de dos maneras:
1) Unos son portadores de oncogenes que alteran la maquinaria celular provocando una proliferación incontrolada de las células hospederas. Se conocen cerca de 20 oncogenes virales asociados con formas particulares de cáncer y cada una tiene un protooncogén correspondiente en las células a las que infectan. 2) Otros retrovirus no portan oncongenes, pero al integrarse al cromosona de la célula receptora alteran la estructura o función de los genes aledaños al lugar de su inserción. Los genes afectados son considerados como protooncogenes potenciales cuya expresión puede alterarse, lo que trae consigo la transformación de las células. |
Los retrovirus se han vinculado con la leucemia de células T en humanos adultos característica del sur de Japón y de las islas del Caribe. Aunque también existen evidencias de que virus cuyo material genético es ADN pueden asociarse con otros padecimientos malignos en el humano, como sucede con el virus de la hepatitis B o con los papilomavirus relacionados con el cáncer de pene en el hombre y el de cuello uterino en la mujer, así como el llamado virus de Epstein-Barr, vinculado con los linfomas de Burkitt y con el cáncer nasofaríngeo.
|
Los virus que insertan su material genético en el
ADN de nuestras células pueden ocasionar problemas de desregulación
en la lectura de la información hereditaria y modificar el comportamiento
celular.
|
¿QUÉ TANTO HA PROGRESADO EL CONOCIMIENTO DE LOS ONCOGENES?
Obviamente el descubrimiento de la existencia de los oncogenes produjo gran entusiasmo y una búsqueda activa de su presencia en las células cancerosas, que aún no termina y ha dado lugar a una multitud de datos no del todo fáciles de descifrar. Se piensa que pronto se requerirá elaborar diagramas con casillas y redes de comunicación, como los que se emplean para describir circuitos electrónicos, sistemas telefónicos o de computación, para lograr establecer los vínculos entre oncogenes, sus productos proteicos, las cadenas bioquímicas que regulan la división y proliferación celular, los cambios en el comportamiento celular y los cánceres resultantes.
A pesar de que todavía no se cuenta con esta información, ya ahora destacan ciertos elementos que se repiten y permiten inferir algunos de los papeles que desempeñan oncogenes y sus consecuencias. Por ejemplo, es notable el hecho de que numerosos oncogenes ya han sido vinculados con algún elemento relacionado con el control de la proliferación celular (Tabla 3). Los productos de tres de los oncogenes enlistados en la tabla (fos, myc y myb) se localizan en el núcleo de las células y parecen participar en la activación de genes que provocan la división celular. En tanto que los productos de los oncogenes restantes se encuentran en la membrana o cerca de ella. Tal es el caso de erb, que es una versión alterada del receptor del factor de crecimiento epidérmico a la que le falta el dominio externo, por lo que se encuentra permanentemente activo. Por su parte, sis controla la síntesis de la cadena B del factor de crecimiento plaquetario, y los restantes están relacionados con enzimas fosforilantes (proteínas cinasas) (Figura 8).
|
TABLA 3. Ejemplos de oncogenes vinculados con el control de la división celular |
||
|
|
||
|
|
|
|
|
|
||
| erb-B | Tirosina cinasa | Receptor en la superficie celular |
| fms | Tirosina cinasa | Receptor en la superficie celular |
| neu | Tirosina cinasa | Receptor en la superficie celular (aún no identificado) |
| ros | Tirosina cinasa | Receptor en la superficie celular (aún no identificado) |
| sis | Factor de crecimiento FCP (subunidad B) | Señal |
| src | Tirosina cinasa | ¿Transducción de señales? |
| abl | Tirosina cinasa | ¿Transducción de señales? |
| ras | ¿Proteína G? | ¿Transducción de señales? |
| fos | ¿? | ¿Efector de la división celular? |
| myc | ¿? | ¿Efector de la división celular? |
| myb | ¿? | ¿Efector de la división celular? |
|
|
||
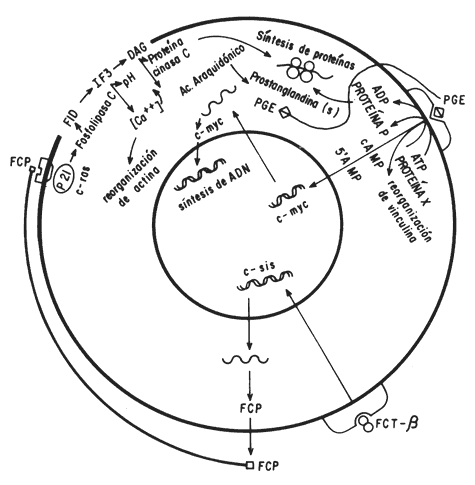
Figura 8. Participación de los factores del crecimiento y productos oncogénicos en la proliferación celular.
En esta figura se puede observar cómo el factor de crecimiento transformante
de tipo beta (FCT- ß) al unirse a su receptor, activa la expresión del oncogén
c-sis, el cual determina la síntesis del factor de crecimiento plaquetario (FCP).
Este factor, a su vez, se une a su receptor e induce la activación de
la fosfolipasa C, probablemente a través de una proteína G (¿p-21 codificada
por el oncogén ras?). La fosfolipasa rompe al fosfatidil inositol difosfato
(FID) liberando al inositol trifosfato (IF3) y al diacilglicerol
(DAG), lo cual conduce a la activación de la proteína cinasa C,
a la formación de ácido araquidónico y prostaglandinas. La prostaglandina E
se une a un receptor e induce la síntesis del adenosín monofosfato cíclico y
la activación del oncogén myc, todo lo cual provoca la síntesis de proteínas,
del ADN y la división celular, junto con la reorganización de las proteínas
del citoesqueleto (actina y vinculina), bajo la influencia del incremento del
calcio citoplásmico y de la elevación del pH intracelular. Por su parte, el
oncogén erb-B determina la síntesis del dominio interno del receptor del factor
de crecimiento epidérmico, el cual es regulado por la acción fosforilante de
la proteína C inducida por FCP.
Se ha descubierto también que no basta con que se active un oncogén en una célula para que ésta adquiera las características de una célula cancerosa, sino que se requiere por lo menos la participación de dos oncogenes. Así, por ejemplo, ni el oncogén Ela ni el Elb del adenovirus pueden inducir tumorigenicidad por separado, pero actuando juntos provocan la transformación maligna de las células; lo mismo ocurre con los oncogenes celulares ras y myc que por sí solos no son suficientes para inducir tumores, lo que sí ocurre cuando se activan ambos.
|
Cada día se aprende algo nuevo sobre los oncogenes,
pero todo parece girar en torno de unos cuantos principios. Así, por
ejemplo, la localización de sus productos proteicos en el citoplasma
o el núcleo parece desempeñar un papel clave en su función, también
sorprende saber que, con unos cuantos genes y dependiendo del orden
en que se activen o de la combinación de oncogenes activos, se pueden
dar modalidades distintas del cáncer.
|
¿QUÉ SE SABE SOBRE LA ACTIVACIÓN DE LOS ONCOGENES?
Se ha mencionado ya que la integración de los virus en el ADN de las células hospederas puede modificar la estructura o función de los protooncogenes, activándolos y convirtiéndolos en oncogenes.
En las células que no han sido infectadas por virus y en las que ocurre la activación anormal de los protooncogenes, se ha visto que ésta puede producirse como resultado de tres mecanismos (Tabla 4):
—Una mutación puntual, que cambia la secuencia de bases en el gen y que se refleja en un cambio en la secuencia de aminoácidos de la proteína oncogénica.
—Una movilización (translocación) del onocogén a otro cromosoma, al que se ubica en vecindad con un gen continuamente activo que lo estimula a manifestarse,
—La multiplicación en el número de copias del oncogén (amplificación).
TABLA 4. Mecanismos de activación de oncogenes de células humanas |
|||
Oncogén |
Mecanismo de activación |
Tipo de tumor |
Porcentaje de tumores |
que lo contienen |
|||
H-ras |
Mutación puntual |
Carcinomas y melanomas |
1-10 |
K-ras |
Mutación puntual |
Carcinomas, leucemias linfoides agudas, rabdomiosarcomas, |
1-15 |
Amplificación |
Cáncer de vejiga y pulmón |
1 |
|
N-ras |
Mutación puntual |
Varias leucemias, carcinomas, melanomas y sarcomas |
1-25 |
c-myc |
Translocación |
Linfoma de Burkitt |
100 |
Amplificación |
Carcinoma de pulmón de células pequeñas, cáncer de colon y mama, leucemia promielocítica |
1-35 |
|
| |
|||
Se ha visto que la activación de los oncogenes ras y neu, mediante mutaciones
puntuales, puede ocurrir en tumores inducidos en animales mediante el empleo
de sustancias cancerígenas o por rayos X (Tabla 5). Así, por
ejemplo, se ha encontrado el oncogén H-ras-1 en carcinomas
de células escamosas inducidos en la piel de ratones por la aplicación de dimetilbenz
(a) antraceno (DMBA),seguida de la administración de ésteres de
forbol, y en cánceres mamarlos producidos durante el desarrollo sexual por N-nitroso-N-metilurea
(NMU). También se ha visto que el oncogén ras puede activarse
en adenomas y carcinomas "espontáneos" en el ratón (cepa B6C3FI
empleada en bioensayos de carcinogénesis).
TABLA 5. Oncogenes en tumores inducidos por carcinógenos químicos |
||||
Porcentaje de |
||||
Especie |
tumores con el |
|||
animal |
Tumor |
Carcinógeno |
Oncogén |
oncogén activado |
Rata |
NMU |
Carcinoma mamario |
H-ras 1 |
86 |
DMBA |
Carcinoma mamario |
H-ras 1 |
23 |
|
DMN |
Carcinoma renal |
K-ras 2 |
40 |
|
ENU |
Neuroblastoma |
neu |
100 |
|
NMU |
Schwanomas |
neu |
70 |
|
NMS |
Carcinomas nasales |
? |
100 |
|
Ratón |
DMBA |
Carcinomas de piel |
H-ras 1 |
90 |
Varios |
Hepatomas |
H-ras 1 |
100 |
|
Rayos X |
Linfomas |
K-ras 2 |
57 |
|
NMU |
Linfomas |
N-ras 1 |
85 |
|
3-MCA |
Fibrosarcoma |
K-ras 2 |
50 |
|
NMU = Nitroso metil urea, |
||||
DMBA = Demitilbenz (a) antraceno, |
||||
DMN = Dimetil nitrosamina, |
||||
ENU = Etil nitroso urea, |
||||
MMS = Metil metano sulfonato. |
||||
3-MCA = 3-Metil colantreno |
||||
|
|
||||
Lo anterior muestra que se puede inducir la activación de oncogenes tanto por la exposición a agentes físicos, químicos y virales, como probablemente por la intervención de factores endógenos que hacen a algunos individuos más propensos a desarrollar "espontáneamente" cáncer.
|
Un gen normal puede convertirse en un gen del cáncer
al ser alterado por agentes ambientales o generados dentro de nuestro
organismo cuando fallan los mecanismos de protección de los que disponemos.
Algunas personas tienen mecanismos de protección ineficientes que las
hacen más propensas a padecer cáncer.
|
¿QUÉ RELACIÓN TIENEN LOS ONCOGENES CON LAS ETAPAS DEL CÁNCER?
Los estudios experimentales de inducción de cáncer en la piel de ratones por la aplicación de compuestos químicos, ha sido una rica fuente de información en lo que se refiere al conocimiento y caracterización de las etapas por las que atraviesa el proceso de carcinogénesis.
En la década de 1940, diversos trabajos mostraron que ciertas sustancias, administradas en una dosis alta, eran capaces de provocar tumores malignos por sí solas, por lo que se les denominó carcinógenos completos. Por el contrario, se observó que una dosis baja de dichas sustancias sólo producía cáncer si se acompañaba de la aplicación repetida de otra sustancia capaz de promover la división celular (agente promotor). En un principio tal hallazgo hizo pensar que el cáncer pasaba por una primera etapa de "iniciación'' (inducida por un agente iniciador), seguida por una segunda etapa de "promoción".
Más tardé se descubrió que si se aplicaba alguna dosis de un agente iniciador y se espaciaba hasta un año la aplicación del promotor, también se producían tumores malignos. Esto llevó a pensar que la célula "guardaba la memoria" del cambio provocado por el iniciador; dicho cambio, entonces, era irreversible y debería corresponder a una mutación, es decir, a una modificación en el material genético de la célula. Se descubrió además que la aplicación de un promotor en ausencia del iniciador o durante un tiempo insuficiente, no era capaz de provocar tumores (Figura 9).

Figura 9. Inducción de tumores por la aplicación de iniciadores y promotores
El estudio de los tumores permitió la identificación de múltiples cambios en el número y estructura de los cromosomas de las células tumorales, a medida que se volvían invasivos. Esto, junto con otros hallazgos, llevó a concluir la existencia de una tercera etapa en el desarrollo del cáncer: la "progresión tumoral", así como a considerar que se requería un segundo acontecimiento genético (probablemente mediado por una alteración cromosómica) para que las células adquirieran verdaderamente características cancerosas (Figura 10).
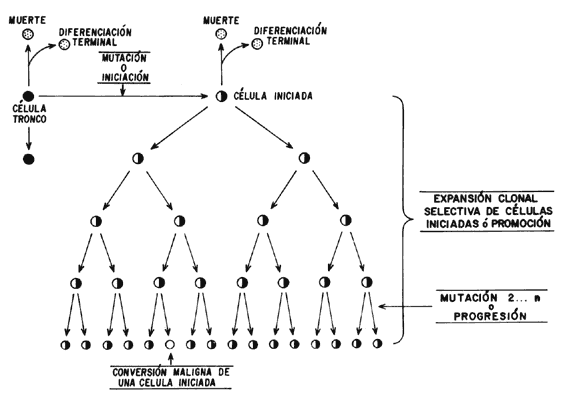
Figura 10. Iniciación, promoción y progresión del cáncer.
Como se mencionó anteriormente, se han aislado oncogenes portadores de mutaciones
puntuales (H-ras1) en tumores provocados por la aplicación
sucesiva de iniciadores (DMBA) y promotores (ésteres de forbol),
lo cual coincide con la hipótesis de que el inicio del cáncer está mediado por
un cambio genético. También apoya esta idea el hallazgo del oncogén H-ras
activado en papilomas, considerados como la fase premaligna de los cánceres
de piel. Numerosas observaciones corroboran la intervención de mutaciones puntuales
en los oncogenes de la familia ras, como paso inicial en diversas formas de
carcinogénesis, aunque no en todas, lo que indica que también otros cambios
pueden iniciar el proceso; de ellos se hablará más adelante.
Algo interesante ha sido encontrar que las mutaciones en el protooncogén ras se dan repetidamente en dos codones (secuencia de tres nucleótidos) específicos, que ocupan las posiciones 12 (Guanina-Guanina-Adenina) y 61 (Citosina-Adenina-Adenina). Esas mutaciones tienen como resultado que en la proteína p2l, codificada por ras, se altere el sitio clave que le permite unirse al GTP, con lo que éste permanece más tiempo unido a ella; como consecuencia final de lo anterior, también se altera la regulación de la división celular.
Sumado a eso, se ha visto que en los carcinomas de piel, que corresponden a la fase de progresión tumoral o transformación maligna de los papilomas, el oncogén ras mutado puede amplificarse. Esto coincide con la propuesta de la producción de un segundo acontecimiento genético, que desencadena la malignización. También se ha observado una estrecha asociación entre la amplificación del oncogén myc y los estados avanzados del cáncer; por ello se considera pues, que dicha amplificación contribuye a la progresión, como sucede en el cáncer del cuello del útero. Los oncogenes ras y myc parecen complementarse uno a otro, y si, como ya se había mencionado, cada uno por separado no induce por sí mismo la transformación maligna completa de las células, ello sí ocurre si ambos son activados.
Si se analiza la figura 8 se puede ver cómo ras y myc toman parte secuencialmente
en una de las cascadas bioquímicas que desencadenan la división celular. Se
ha visto que, curiosamente, agentes promotores como los ésteres de forbol y
en particular el TPA (12-0-tetradecanoilforbol 13-acetato) parecen
estimular la proliferación celular de manera muy similar a como lo hacen los
factores de crecimiento y poseen receptores en la membrana celular de fibroblastos
y células epidérmicas del ratón. El TPA, al igual que el factor
de crecimiento plaquetario, estimula la activación de una proteína cinasa C,
otro elemento clave en la regulación de la división celular que aparece ubicado
en la cadena de sucesos que se describen en la figura 8.
|
Diversas pruebas indican que las fases por las que
atraviesa el proceso de carcinogénesis se asocian con cambios peculiares
en distintos grupos de oncogenes; se está investigando el significado
de esos cambios.
|
¿QUÉ FACTORES DENTRO DEL ORGANISMO PUEDEN FAVORECER LA ACTIVACIÓN DE ONCOGENES?
La investigación de los procesos bioquímicos normales de las células ha permitido descubrir que incluyen la producción de moléculas muy reactivas (radicales libres) que pueden ocasionar daños celulares y provocar lesiones en el ADN.
Por ejemplo, el oxígeno y el hierro, dos elementos esenciales de la vida, están
implicados en la formación de radicales sumamente tóxicos. El oxígeno es fácilmente
reducido para dar lugar a la formación del anión superóxido ( ![]() ) y enseguida a peróxido de hidrógeno (
) y enseguida a peróxido de hidrógeno ( ![]() ), ambos radicales altamente tóxicos son producidos por los leucocitos neutrófilos
como un mecanismo natural para combatir las infecciones microbianas. El hierro
inorgánico es capaz de catalizar la conversión del anión superóxido y del peróxido
de hidrógeno en radicales mucho más tóxicos como los radicales hidroxilos (OH).
Estos se han asociado con la peroxidación de los lípidos de las membranas y
la inducción de rompimientos cromosómicos y alteración de las bases del ADN.
), ambos radicales altamente tóxicos son producidos por los leucocitos neutrófilos
como un mecanismo natural para combatir las infecciones microbianas. El hierro
inorgánico es capaz de catalizar la conversión del anión superóxido y del peróxido
de hidrógeno en radicales mucho más tóxicos como los radicales hidroxilos (OH).
Estos se han asociado con la peroxidación de los lípidos de las membranas y
la inducción de rompimientos cromosómicos y alteración de las bases del ADN.
Al conocerse que agentes inflamatorios pueden actuar como promotores en el proceso de carcinogénesis, se ha pensado que esto puede ser el resultado de la liberación de radicales libres, como ocurre en los procesos inflamatorios en los que intervienen los leucocitos neutrófilos antes citados. A esto se suman las observaciones de rompimientos cromosómicos y alteraciones oxidativas del ADN en presencia de agentes promotores.
Afortunadamente, los seres vivos han desrrollado mecanismos para protegerse de los efectos indeseables de los radicales libres; entre ellos se encuentran las enzimas superóxido desmutasa y catalasa, que catalizan la conversión del anión superóxido a peróxido y la de éste en agua y oxígeno. Además, existen otros componentes celulares o sustancias ingeridas con los alimentos que atrapan a los radicales libres, como el glutatión, los flavonoides, retinoides y la vitamina C; por lo que la nutrición se vuelve un elemento esencial contra las lesiones autooxidantes pues aporta dichos elementos protectores. Coinciden en destacar la importancia de la alimentación las observaciones que demuestran que se puede inhibir la liberación de radicales libres, la expresión de oncogenes y la promoción con el empleo de la vitamina C, los retinoides, el aceite de cebolla y los inhibidores de proteasas.
Se sospecha que algunos individuos tienen deficiencias en los mecanismos de defensa en contra de los radicales libres, ya sea por problemas hereditarios que afectan sus sistemas enzimáticos o por problemas nutricionales, y que esto pueda ocasionarles una mayor susceptibilidad a sufrir cáncer tanto por la agresión de radicales endógenos como por la producida por agentes ambientales oxidantes.
|
Dentro de nuestro organismo se llevan a cabo procesos
en los que se generan moléculas sumamente reactivas que, de no ser estabilizadas,
pueden ocasionar daños genéticos o estímulos proliferativos, los cuales
provocan el desarrollo de cáncer en individuos desprovistos de defensas
en contra de ellas.
|
¿EXISTEN PROTEÍNAS QUE SUPRIMEN EL COMPORTAMIENTO ANORMAL DE LAS CÉLULAS CANCEROSAS?
Las células cancerosas se distinguen, además de su proliferación autónoma y continua, por alteraciones relacionadas con la diferenciación celular. Ya se describió cómo en las etapas iniciales del proceso de carcinogénesis las células muestran cambios que las hacen diferentes de las células del tejido al que pertenecen y conducen a lo que se conoce como metaplasias, displasias, proliferaciones focales y papilomas. Esto ha llevado a pensar que la proliferación de estas células premalignas resulta también de un impedimento para seguir sus procesos normales de diferenciación.
Al investigar los mecanismos de diferenciación celular, se ha descubierto que están basados en sistemas complejos de señales y cascadas bioquímicas, similares a las descritas con respecto de la regulación de la proliferación celular; se ha confirmado pues que intervienen primeros mensajeros (como los factores de crecimiento) y segundos mensajeros, o sea que las mismas proteínas reguladores deben intervenir en los dos procesos. Ahora se estudia a qué obedecen los diferentes resultados y en qué condiciones una señal lleva a la célula a dividirse y en cuáles otras induce su diferenciación.
El factor de crecimiento transformante tipo beta (FCT-b
es un caso particularmente ilustrativo de la doble función de las proteínas
reguladoras a las que se hace referencia (Figura 8), FCT-b
funciona como un estimulador del crecimiento sólo en ciertas células fibroblásticas,
a las que parece inducir a sintetizar, secretar y responder en forma autocrina
a un factor de crecimiento (factor de crecimiento plaquetario codificado por
el protooncogén FCT-b,
que las lleva a dividirse. Existen pruebas de que este factor interviene en
los procesos de cicatrización, en los que es necesaria la proliferación de fibroblastos
para cerrar heridas. Sin embargo, su función más usual, pero menos conocida
desde el punto de vista mecánico, es la inhibición de la división celular. En
este caso también parece provocar la producción y respuesta autocrina a una
proteína inhibitoria, la cual bloquea más eficientemente a células normales
que a cancerosas; esto parece indicar que las células cancerosas han sufrido
cambios que las hacen menos susceptibles a los inhibidores del crecimiento que
produce el organismo como parte de los mecanismos de homeostasis.
Las células precursoras de los componentes de la sangre (hematopoiéticas) constituyen un modelo para el estudio de los mecanismos que llevan a las células a decidir entre proliferar y diferenciarse, pues se ha visto que los protooncogenes que inducen en ellas la división también dan lugar a un bloqueo aparente en la diferenciación terminal.
El conocimiento de los mecanismos por los cuales las células cancerosas bloquean su capacidad de diferenciarse, así como de las proteínas que actúan en cada tipo particular como señales para inducir la diferenciación, se espera lleve a descubrir el modo de inducir a las células tumorales a proseguir su diferenciación normal. En este aspecto el modelo de las células hematopoiéticas ha aportado resultados esperanzadores, por lo menos en los ensayos hechos en cultivos celulares.
|
Las células no parecen necesitar de recursos y mecanismos
distintos para realizar funciones diferentes como proliferar o diferenciarse,
sino que su comportamiento depende de la combinación, secuencia e intensidad
de la intervención de unas cuantas moléculas clave, lo cual hace pensar
que se puede reorientar el comportamiento de las células cancerosas
utilizando mensajes adecuados.
|
¿EXISTEN GENES ESPECÍFICOS QUE SUPRIMEN EL COMPORTAMIENTO CANCEROSO DE LAS CÉLULAS?
En experimentos de fusión de células normales con otras de tumores animales o transformadas por virus oncogénicos, se ha logrado suprimir la tumorigenicidad de estas últimas. Esto ha hecho suponer la existencia, en las células normales, de genes que pueden suprimir la manifestación de las características cancerosas provocadas por los oncogenes.
Algo similar a lo anterior se ha descubierto en el estudio de las células del cáncer del cuello del útero, que, como se señaló antes, está asociado con la infección por virus oncogénicos. En este caso particular, la supresión del comportamiento tumoral por la fusión con células normales hablaría en favor de que en las células humanas existen mecanismos de defensa ancestrales que las protegen de los efectos adversos de los virus que producen cáncer. Es posible que la mutación o inactivación de los genes supresores sea lo que tiene como consecuencia el que los oncogenes virales queden libres de manifestarse. La observación de la existencia de un mayor riesgo de desarrollar el cáncer de útero en las mujeres fumadoras hace creer que los agentes mutagénicos presentes en el tabaco pudieran ser los responsables de la inactivación de los genes supresores.
|
Todo parece indicar que en el material hereditario
de nuestras células está contenida la información que, de acuerdo con
las circunstancias, puede favorecer o impedir el desarrollo del cáncer.
|
¿PUEDE EL CÁNCER HEREDARSE DE PADRES A HIJOS?
El descubrimiento de familias en las que numerosos miembros padecieron cáncer a lo largo de varias generaciones ha llamado la atención desde hace mucho tiempo. Entre 1866 y 1869, el médico francés P. Broca describió a una familia en la que murieron de cáncer la madre, sus cuatro hijas y ocho de los hijos de éstas. El médico inglés W. Williams refiere una experiencia similar en 1898, en la que una mujer afectada por cáncer uterino sufrió la pérdida de dos hermanas, de su madre, una tía y su abuela por línea materna, todas ellas por el mismo tipo de cáncer. Estudios semejantes en países como Estados Unidos han notificado la muerte por cáncer de 13 individuos de una misma familia a lo largo de cinco generaciones, en diez de los cuales el cáncer atacó el intestino grueso.
Estas observaciones sugirieron un modo de transmisión hereditario en esas familias. Se calcula que los cánceres de esta índole constituyen alrededor del 1% del total de casos de esta enfermedad.
Es preciso hacer notar que en los países en los que el cáncer ocupa uno de los primeros lugares como causa de enfermedad y muerte no es raro ver aparecer en una misma familia más de un caso de este padecimiento. Esto es lo que ocurre en Estados Unidos, en donde 20% de las muertes se deben al cáncer y uno de cada cinco individuos fallece por esta enfermedad. A pesar de ello, varias características permiten distinguir los casos de transmisión hereditaria, entre las cuales se encuentran las siguientes:
—Varios familiares afectados por el mismo tipo de cáncer.
—Un número mayor de casos que en la mayoría de las familias.
—Forma de transmisión hereditaria de tipo mendeliano.
—Aparición de múltiples tumores en el mismo individuo.
—Asociación entre casos de cáncer, padecimientos hereditarios, malformaciones al nacimiento y abortos en la misma familia.
A la fecha, se han identificado alrededor de 50 tipos de cáncer hereditario como los que aparecen listados a continuación y se sospecha que existen muchos más:
|
|
|
|
|
|
|
|
|
| Carcinoma neviode de células basales |
|
| Poliposis y carcinoma de colon |
|
| Adenomatosis endocrina múltiple |
|
| Feocromocitoma y cáncer medular de tiroides |
|
| Tilosis con cáncer esofágico |
|
|
|
|
Se ha observado en el caso del cáncer de mama que puede existir una propensión en ciertas familias a que más de un miembro de sexo femenino de la familia desarrolle la enfermedad. Por ello, y como una medida para detectar oportunamente el inicio de un tumor, se recomienda que mujeres cuyas madres, tías, abuelas maternas o hermanas hayan tenido cáncer de mama vigilen continuamente sus senos para detectar nódulos precursores.
|
La existencia en el árbol genealógico de una familia
de múltiples casos de cáncer, sobre todo de un mismo tipo, debe alertar
sobre la existencia de una propensión hereditaria, para establecer vigilancia
médica de los individuos sanos.
|
¿QUÉ RELACIÓN TIENEN LOS CÁNCERES HEREDITARIOS CON LOS ONCOGENES?
Las observaciones realizadas por el doctor A. G. Knudson colaboradores en los pacientes afectados por retinoblastoma han permitido vislumbrar cómo se originan los cánceres hereditarios y su posible relación con los oncogenes.
El retinoblastoma es un tumor de los ojos que se origina en las células de la retina y los individuos que lo padecen pueden pertenecer a dos grupos (hereditario y no hereditario). En uno de dichos grupos, el cáncer hace su aparición antes del primer año de vida, frecuentemente es bilateral, o sea que afecta a ambos ojos, y los individuos que lo padecen tienen, por lo general, un progenitor también afectado por la misma forma de cáncer (el tumor puede ser extirpado y los individuos sobreviven y pueden reproducirse, aunque transmiten la propensión a desarrollar el tumor a sus descendientes).
El otro grupo de individuos presenta el tumor más tarde, pero antes de los cinco años de edad (después de esa edad los retinoblastos dejan de dividirse y son menos susceptibles a transformarse en células cancerosas), además de que es común que sólo tengan un tumor en un ojo y no tienen parientes afectados.
Lo anterior hizo suponer al doctor Knudson que los casos de retinoblastoma hereditario deberían ser causados por la transmisión de un gen recesivo, que no se manifiesta sino hasta que su homólogo normal (todos los genes de las células, salvo algunos de los cromosomas sexuales, se presentan en pares) sufre una mutación que lo inactiva o se pierde. En los individuos que no presentan la forma hereditaria del tumor, Knudson supuso que se requerían dos mutaciones sucesivas para inactivar a los dos genes y provocar el tumor, por ello era más probable que tuvieran tumores sólo en un ojo.
Estos descubrimientos coinciden con algunos de los planteamientos sobre el proceso de carcinogénesis hechos anteriormente, pues en la evolución del retinoblastoma también se da por etapas; las células sufren primero un acontecimiento genético que las prepara (iniciación) para responder después, tras una segunda mutación, con un comportamiento proliferativo anormal. En este caso se interpretó que el papel que desempeñan los genes participantes se parecía al de un posible antioncogén que mientras se encuentra en su versión normal suprime la expresión de los oncogenes, pero al inactivarse lo deja libre para manifestarse. Algo similar se ha encontrado en otro tumor de tipo hereditario, el tumor de Wilms cuyo gen participante se localiza en el cromosoma 11, y en el cáncer de mama de origen familiar.
No se sabe aún qué relación existe entre estos antioncogenes y los genes supresores del comportamiento canceroso de las células, a los que se hizo alusión anteriormente. Actualmente se ha propuesto y se investiga la posibilidad de que oncogenes, genes supresores y antioncogenes sean en realidad una misma familia de genes y que su diferente comportamiento obedezca a la modulación de su manifestación.
|
Poco a poco la información de distintos tipos de estudios
parece encajar como en un rompecabezas señalando a los genes responsables
del desarrollo de procesos cancerosos y relacionándolos con los mecanismos
que regulan la proliferación y diferenciación celular.
|
¿EXISTEN PADECIMIENTOS HEREDITARIOS QUE AUMENTEN EL RIESGO DE PADECER UN CÁNCER?
La existencia de una asociación entre diversos padecimientos hereditarios y el desarrollo de tumores o leucemias constituye un hecho de particular interés, sobre todo en aquellos casos en los que se constata, además, una fragilidad cromosómica que lleva a rearreglos genéticos diversos y en los que se presentan mutaciones frecuentes, antes de que hagan su aparición padecimientos malignos. Ejemplo de ellos son el síndrome de Bloom, la anemia de Fanconi, y la Ataxia telangiectasia. Otro padecimiento hereditario igualmente interesante es el conocido como Xeroderma pigmentosum, en el cual los individuos afectados son propensos a desarrollar cáncer de piel en virtud de su extrema susceptibilidad a la luz ultravioleta del sol y a una deficiencia en su capacidad de reparar el daño que provoca dicha radiación en el material genético de las células de la piel. Las células de estos individuos al ser sometidas a distintos agentes físicos, químicos o virales, muestran una mayor frecuencia de alteraciones en su material genético que las células normales. Tanto los cánceres de tipo hereditario como este tipo peculiar de asociaciones entre enfermedades hereditarias, vulnerabilidad a agentes ambientales, lesiones genéticas y padecimientos malignos, constituyen modelos a investigar para desentrañar la relación que existe entre la herencia, el ambiente y el cáncer.
| Todo agente externo o interno que provoque lesiones genéticas en las células del organismo conlleva el riesgo de producir un cáncer, particularmente si coincide con estímulos a la proliferación de las células alteradas. |
¿EXISTEN CARACTERÍSTICAS HEREDITARIAS QUE INCREMENTEN LA SUSCEPTIBILIDAD A LOS CARCINÓGENOS AMBIENTALES?
Un gran número de sustancias potencialmente tóxicas que ingresan a nuestro organismo son transformadas por enzimas presentes en distintos órganos entre los que sobresale el hígado; lo mismo sucede con aquéllas capaces de inducir cáncer. En algunos casos las sustancias no son tóxicas sino hasta después de ser metabolizadas, por lo que se dice que las enzimas las activan; en otros casos son inactivadas por procesos enzimáticos de destoxificación.
Los seres humanos difieren en su capacidad de llevar a cabo tales procesos, ya que pueden contar con enzimas que presentan distinta actividad (isoenzimas) como resultado de los cambios genéticos que han ido ocurriendo a lo largo de la evolución. Ciertos individuos deficientes en ciertas enzimas biotransformadoras pueden ser menos susceptibles a carcinógenos químicos que requieren activación metabólica. Por el contrario, otros pueden ser más vulnerables por no contar con mecanismos eficientes de destoxificación de carcinógenos.
|
Entre los mecanismos moduladores de nuestra respuesta
a la agresión de sustancias cancerígenas se encuentran las enzimas capaces
de metabolizarlas.
|