VI. FARMACOCINÉTICA
EL PASO DE FÁRMACOS A TRAVÉS DE BARRERAS BIOLÓGICAS
PARA QUE UN FÁRMACO ACTÚE es necesario que llegue a su sitio de acción. Para ello, la sustancia tiene que absorberse, esto es, alcanzar el compartimiento acuoso del organismo. Excepto la piel y algunas mucosas, en todos estos mecanismo participa la sangre. Así, la distribución del fármaco dentro del cuerpo puede variar de acuerdo con el flujo sanguíneo o la vascularización regional de cada tejido u órgano, y la cantidad de droga que cada tejido reciba depende de la concentración del fármaco en la sangre. A su vez, la magnitud del efecto varía por la velocidad con la que el fármaco penetra al tejido hasta alcanzar niveles suficientes.
Un fármaco puede administrarse por vía enteral o por vía parenteral, inyectarse
directamente al espacio intravascular o ser depositado en sitios fuera de este
espacio, para su absorción gradual. El sistema gastrointestinal es el sitio
habitual para ello, aunque las vías pulmonar (por inhalación), subcutánea e
intramuscular son otras opciones (véase la figura VI.I).
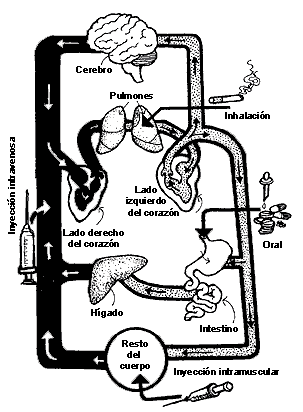
FIGURA VI.I. Esquemas de las vías de
administración y distribución de las drogas. Diferentes vías de administración
de fármacos enterales o parentales (inhalación, intramuscular, intravenosa).
Una vez dentro del cuerpo, el fármaco se distribuye por medio de la sangre a
los diversos órganos y sistemas.
En el cuadro VI.I se resumen las principales vías de administración,
sus ventajas y desventajas.
C
UADRO VI.I. Vías de administración
| |
|||
| Vía |
Ventajas |
Desventajas |
Ejemplos |
| |
|||
| ENTERAL | |||
| Oral | Fácil, segura,conveniente | Absorción limitada o errática de algunas drogas; posibilidad de inactivación hepática | Analgésicos, sedantes e hipnóticos, etcétera |
| Sublingual | Inicio rápido del efecto. No se inactiva en el hígado | El fármaco debe absorberce en la mucosa oral | Nitroglicerina |
| Rectal | Opción de la vía oral. Efectos locales en la mucosa rectal | Absorción pobre o incompleta. Riesgo de irritación rectal | Laxantes, supositorios y otros |
| PARENTAL | |||
| Inhalación | Inicio rápido. Aplicación directa en alteraciones respiratorias. Gran superficie de absorción | Riesgo de irritación tisular. Problemas de dosificación | Anestésicos generales, agentes antiasmáticos |
| Inyección (SC, IV, IP, intratecal*) | Administración a órganos blanco. Inicio rápido | Riesgo de infección. Dolor. Imposibilidad de recuperar la droga. Sólo fármacos solubles | Insulina, antibióticos, drogas anticancerígenas, narcóticos |
| Tópica | Efectos locales sobre la superficie de la piel | Sólo eficaz en capas superficiales de la piel | Ungüentos, cremas, gotas nasales y oculares, preparaciones vaginales |
| |
|||
SC=subcutánea; IV= intravenosa; IP= intraperitoneal.
* Se refiere a la administración en el canal raquídeo
Para que una sustancia atraviese las membranas celulares es condición esencial que se encuentre en forma libre, es decir, que no esté unida a otras moléculas. En la sangre, la albúmina representa una proteína con múltiples sitios de unión para fármacos. Mientras éstos se mantengan unidos a la albúmina no podrán abandonar el torrente sanguíneo y, por lo tanto, no llegarán a sus sitios de acción. Por otra parte, los fármacos, a su vez, competirán con otras moléculas endógenas contenidas en la sangre (p ejem., hormonas, bilirrubina, vitaminas, iones, etc.) por los sitios de transporte, con consecuencias potencialmente peligrosas de acumulación.
El paso de fármacos a través de las barreras biológicas está condicionado por las características fisicoquímicas de la sustancia. En particular, del tamaño o peso molecular; grado de ionización (carga eléctrica) y liposolubilidad (capacidad de disolverse en las grasas). Así, una sustancia pequeña, poco ionizada y muy liposoluble atraviesa rápidamente las membranas celulares. Tal es el caso de la mayoría de los anestésicos volátiles, agentes broncodilatadores o solventes orgánicos.
La transferencia (translocación) de fármacos a través de barreras membranales puede realizarse por filtración, difusión, transporte activo, pinocitosis o fagocitosis (procesos en los que la célula envuelve e introduce moléculas a su interior). La diferencia de estos procesos depende del tamaño de la droga que se transporte, su solubilidad y la necesidad de acarreadores membranales. Para la filtración y la difusión, la velocidad de transferencia depende también del gradiente de concentración del fármaco en ambos lados de la membrana. En el caso del transporte activo, una sustancia puede ser introducida al espacio intracelular independientemente de su tamaño o liposolubilidad; sin embargo, en esta situación se requiere de cierta especificidad estructural; recordemos que este transporte activo es un mecanismo saturable y dependiente de energía.
Finalmente, es necesario considerar la biodisponibilidod, entendida como la facilidad con la que un fármaco se incorpora a sus sitios de acción; aquí se incluye la presentación farmacéutica en la que se ofrece el medicamento. La absorción no es la misma para una tableta que para una cápsula, que para una preparación de liberación prolongada. En este último caso, la sustancia activa se halla incluida en una matriz de degradación lenta que va liberando gradualmente el principio activo, y como la dosis que se administra representa varias dosis únicas, existe el peligro potencial de una liberación masiva del fármaco contenido en la preparación y los consecuentes efectos tóxicos por sobredosis.
En relación con la distribución del fármaco, una vez que alcanza el espacio intravascular, es necesario tomar en cuenta su volumen aparente de distribución (Vd), o sea, el volumen fluido en el que el fármaco se distribuye, puesto que es un índice de la compartimentalización de la sustancia. Un fármaco con Vd elevado es una sustancia que se almacena o secuestra en algún compartimiento del organismo, por lo que tendrá un potencial de toxicidad por acumulación. El Vd es diferente entre niños y adultos, y entre sujetos sanos y enfermos.
Así, la distribución de una fármaco determinará en parte la latencia, intensidad y duración de la actividad biológica del fármaco. Existen varios factores que pueden afectar el Vd: la afinidad del fármaco por las moléculas transportadas por la sangre, el flujo sanguíneo regional, la afinidad por los componentes de los tejidos, las barreras especiales (p. ejem., placenta, cerebro), factores fisiológicos (p. ejem., ritmos biológicos, pH, glicemia, etc.), patológicos (p. ejem., inflamación o edema, diarrea, fiebre, etc.) y farmacológicos (p. ejem., interacción con otras sustancias).
Mencionemos los casos especiales del paso de fármacos al sistema nervioso y al feto.
En el caso del cerebro y médula espinal, muchas sustancias pasan de la sangre al líquido cefalorraquídeo (LCR) de los ventrículos cerebrales. El LCR se forma cuando la sangre pasa a través de los plexos coroideos donde células especializadas filtran y cambian su composición. Así, el LCR transporta sustancias alimenticias, hormonas o productos de desecho a los sitios más profundos del SNC, allí donde los vasos sanguíneos son demasiado pequeños o insuficientes para mantener la función de esas estructuras.
En cuanto al feto, la distribución de todo tipo de sustancias es a través de los vasos umbilicales formados por tejido placentario. La placenta es un órgano en sí: tiene una estructura anatómica definida, con capacidad de filtrar y metabolizar sustancias provenientes de la sangre materna (incluyendo fármacos), así como de producir hormonas necesarias para el proceso de la gestación, además, sirve de barrera entre el ambiente de la madre y el feto; y las drogas que atraviesan la barrera placentaria pueden afectar al feto a veces en forma irreversible.
La eliminación de un fármaco se efectúa por medio del metabolismo, el almacenamiento y la excreción. Todos estos procesos tienden a disminuir los niveles extracelulares del fármaco. El proceso más frecuente es el de la excreción a través de los riñones, sistema biliar, intestino y, en ocasiones, los pulmones.
La excreción renal de fármacos representa el mecanismo predominante de eliminación. Las diferentes porciones de la nefrona, unidad funcional del riñón, realizan funciones de filtración, secreción y excreción diferencial las cuales pueden alterarse por cambios fisiológicos o patológicos. Así, la acidificación de la orina tiene como consecuencia una mayor ionización del fármaco y aumento en la eliminación de sustancias con pH —grado de acidez— elevado (bases débiles). Una aplicación de este principio sería administrar bicarbonato (es decir, un álcali) para acelerar la eliminación de barbitúricos (que son ácidos) en casos de intoxicación. En otras palabras, restablecer el equilibrio ácido-básico.
El metabolismo (biotransformación) de fármacos se realiza, en gran parte, en el hígado. En este órgano (sistema microsomal) hay reacciones químicas que convierten el fármaco en una sustancia menos soluble y más ionizada, por lo tanto, menos absorbible y menos reutilizable (p. ejem., por reabsorción intestinal a partir de la bilis) aunque puede darse el caso de una transformación metabólica necesaria para que ocurra el efecto biológico. Se habla entonces de un proceso de bioactivación. El metabolismo medicamentoso puede inhibirse o estimularse debido a enfermedades sistémicas y locales, malformaciones o exposición previa a otros fármacos. Por ejemplo, en las fases iniciales del alcoholismo existe mayor resistencia al fármaco por inducción enzimática y aumento del metabolismo, mientras que en las fases cirróticas hay mayor sensibilidad al alcohol por pérdida de unidades funcionales hepáticas.
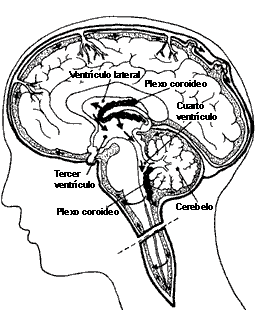
FIGURA VI.2. Producción y distribución del
líquido cefalorraquídeo. Se produce en los plexos coroideos, situados
en los ventrículos cerebrales, desde donde se distribuye a todo el sistema interventricular
y alrededor del SNC.
El ritmo de absorción y eliminación de un fármaco depende de los procesos citados anteriormente y determina la frecuencia de administración del medicamento. La farmacocinética integral utiliza el concepto de vida media (el tiempo necesario para que la concentración sanguínea del fármaco se reduzca a la mitad). Cuando se administra un fármaco se trata de establecer una concentración terapéutica en los fluidos biológicos. Esta concentración eficaz es una propiedad característica del fármaco sobre la cual no tenemos control. Si el nivel de la droga en el suero es insuficiente, la respuesta deseada no ocurre. Si el nivel es más elevado, aparecen signos de toxicidad. Los horarios de dosificación comprenden dos variables: la magnitud de la dosis única y la frecuencia con que se administra (intervalo entre las dosis). Las fluctuaciones de los niveles séricos que pueden observarse entre las administraciones son determinadas por varios factores: para un ritmo dado de eliminación, mientras más rápida es la absorción, más grande es la fluctuación. Si la absorción es rápida, los niveles sanguíneos se elevan al principio, pero disminuyen también relativamente rápido y viceversa.
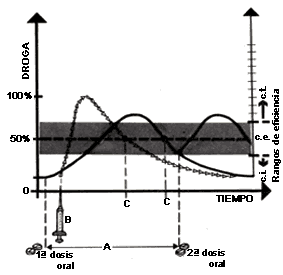
GRÁFICA VI.I. Niveles del fármaco vs. tiempo.
Aquí se representa la evolución temporal de las concentraciones sanguíneas del
fármaco administrado por vía intravenosa (curva punteada) o por vía oral (curva
continua). En la escala de la izquierda se observan los porcentajes de la concentración
sanguínea del fármaco y en la de la derecha la escala de eficacia, indicando
el rango de concentraciones efectivas (c. e.) o terapéuticas (franja sombreada),
así como las concentraciones insuficientes (c. i.) y las tóxicas (c. t.) del
fármaco. La administración intravenosa produce niveles máximos en tiempos cortos,
mientras que la administración oral produce niveles del fármaco que aumentan
más lentamente y que alcanzan concentraciones menores. El intervalo entre las
tomas, indicado con la letra A, está determinado por la vida media del fármaco,
representado por la letra C, señala el tiempo en el que la concentración sanguínea
de la droga disminuye a la mitad (50%).

![[Anteior]](../img/prevsec.gif)
![[Previo]](../img/parntsec.gif)
![[Siguiente]](../img/nextsec.gif)