VII. FARMACODINAMIA
EN GENERAL, los efectos farmacológicos se deben a la interacción entre el medicamento y los componentes específicos del organismo llamados receptores. Los receptores son macromoléculas que pueden estar localizadas en la membrana celular o en el espacio intracelular, y se combinan con el fármaco para producir una reacción química cuya consecuencia es que modifica la función celular. Por lo tanto, para que el efecto biológico aparezca, debe ocurrir primero la unión del fármaco con su receptor. Esta interacción sucede por el establecimiento de uniones químicas, eléctricas o nucleares entre las partes activas de ambas moléculas. Mientras más fuerte sea la unión (p. ejem., es el caso de la unión covalente) más tiempo persiste el efecto farmacológico. En términos prácticos, esto puede significar la irreversibilidad del efecto. Existen otros tipos de unión que participan en la interacción fármaco-receptor, como los enlaces iónicos, los puentes de hidrógeno y los llamados enlaces de Van der Waals (uniones en las que intervienen fuerzas nucleares).
La fuerza de enlace que mantiene combinado a un fármaco con su receptor se debe a la operación concertada de numerosos enlaces de las variedades antes mencionadas. Una vez que la molécula del fármaco es llevada cerca de la superficie del receptor (por procesos de distribución y difusión indicados anteriormente), la agitación térmica aleatorizada produce colisiones múltiples entre las dos superficies. Dado que tanto la droga como el receptor se encuentran rodeados de moléculas de agua (todas nuestras células están rodeadas de agua) es necesario que se establezcan enlaces hidrofóbicos (que tienen aversión por el agua), para que ocurra la interacción. Dicho de otro modo, es necesario eliminar esas moléculas de agua para que los demás grupos químicos puedan reaccionar entre sí. En general, la interacción fármaco-receptor es reversible por disociación de ambas partes, de acuerdo con la ley de acción de masas.
Habíamos mencionado la ionización de un fármaco como factor que influye en el paso a través de membranas. Esta ionización también afecta la interacción entre el fármaco y su receptor.
La teoría nos dice que la magnitud de una respuesta está determinada por el número de receptores ocupados. Se asume que una molécula de cualquier agonista (sustancia que tiene efectos "positivos") que ocupa un sitio receptor hace la misma contribución cuantal a la respuesta total que cualquier otro agonista. Dicho de otra manera, se consideran como enteros —pensando en las fracciones comunes— las unidades que se cuentan cuando se suman o se restan los efectos de agonistas o antagonistas, respectivamente. Sin embargo, hay ocasiones en que varios agonistas que aparentemente actúan en el mismo receptor producen respuestas máximas de magnitud diferente. A partir de estas observaciones se modificó la teoría del receptor adjudicando a cada fármaco dos propiedades independientes relacionadas con su combinación con el receptor: afinidad (tendencia de un fármaco a establecer un complejo o unión estable con el receptor) y eficacia o actividad intrínseca. Esta última describe la eficacia biológica del complejo fármaco-receptor. Se considera que las dos propiedades no son relativas: un agonista y un antagonista pueden tener la misma afinidad por un receptor, pero el primero tiene gran eficacia y poca el segundo. El concepto de eficacia consiste en que agonistas parciales (en el ejemplo que dimos de las fracciones comunes, de sustancias que no aportan enteros sino fracciones del efecto) pueden tener propiedades antagonistas si se les compara con agonistas más potentes.
Mencionemos finalmente los efectos alostéricos, aquellos que ocurren a distancia del receptor y que se manifiestan a nivel de la configuración de la molécula. Puesto que los receptores son proteínas, éstas tienen una estructura terciaria (la forma tridimensional de la molécula) que puede alterarse por varias causas. Por ejemplo, un fármaco que modifica la unión entre un átomo de azufre y otro (en general existen varios de ellos en cada proteína) puede cambiar toda la forma de la molécula. Los llamados radicales libres, por tener gran capacidad de oxidación, pueden modificar la función de la membrana celular, y volverla más permeable a sustancias potencialmente tóxicas del medio extracelular que pueden dañar a la célula. Estos radicales libres son elementos extremadamente reactivos con las proteínas de la membrana celular, que pueden desagregarla, y que se han relacionado con una de las teorías sobre el envejecimiento, la cual postula que éste se debe a la acumulación de radicales libres.
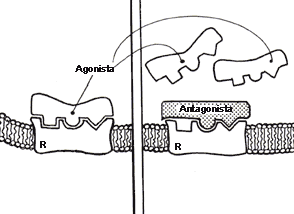
FIGURA VII.I. Receptor: afinidad vs. eficacia.
El receptor (R) localizado en la membrana celular es capaz de reconocer
fármacos con una configuración adecuada, en este caso, un cuadrado, un semicírculo
y un triángulo. Ambos son capaces de ocupar el sitio receptor, es decir, tienen
afinidad por sólo un sitio que contiene las tres formas geométricas, capaz de
producir un efecto farmacológico, eficaz. El antagonista, al ocupar el sitio,
impide que moléculas agonistas actúen en el receptor.
El efecto farmacológico puede resultar de la puesta en marcha de una cascada
de reacciones intracelulares iniciadas desde la superficie celular. En muchos
casos, los efectos intracelulares resultan de la activación, iniciada por la
ocupación del receptor, de los segundos mensajeros, moléculas capaces
de fosforilar proteínas o modificar los estados energéticos de proteínas específicas.
Estos segundos mensajeros se han identificado como derivados de nucleótidos
cíclicos de adenina y guanina, los cuales constituyen familias con diversas
acciones. Las modificaciones de los niveles intracelulares de calcio desempeñan
un papel crítico en estas reacciones.
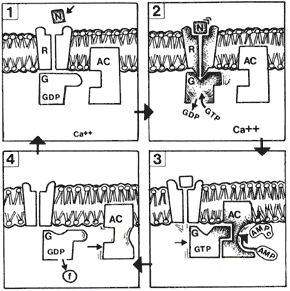
FIGURA VII.2. Mecanismos de transducción y segundos
mensajeros. Los efectos de un neurotransmisor o de un fármaco pueden resultar
de la activación de sistemas que no están ligados directamente a canales iónicos
(véase las figuras V.7 y V.8).
En este caso, la ocupación del receptor (R) induce una cascada de
eventos intracelulares donde participa la adenilato-ciclasa (AC),
la familia de proteínas G, incluida la guanosín difosfato (GDP) y
la guanosín trifosfato (GTP), el calcio (Ca++ ) y el adenosín
monofosfato (AMP). En el cuadro I se representa el sistema
en estado de reposo; el cuadro 2 muestra la ocupación del receptor
por el neurotransmisor (N), provocando que el GTP reemplace
al GDP y el aumento de la concentración intracelular de calcio. Estos
cambios provocan que la proteína G active la adenilato ciclasa (cuadro 3)
y ésta convierta el AMP a su forma cíclica (AMPc),
la cual es capaz de activar o fosforilar otras proteínas intracelulares. Este
AMPc es el segundo mensajero. La hidrólisis del GTP,
produciendo GDP y fósforo inorgánico (P) hace que el sistema retorne
al estado de reposo (cuadro 4).
Existen acciones farmacológicas que no son mediadas directamente por receptores, debidas a efectos inespecíficos; por ejemplo, los diuréticos osmóticos como el manitol o el glicerol, que aumentan la eliminación urinaria porque arrastran moléculas de agua; o los antiácidos, que actúan contrarrestando directamente el exceso de ácido en el estómago, sin interactuar con receptores membranales, o perturbaciones generales de la membrana celular (p. ejem., los anestésicos volátiles), interacciones del fármaco con iones o moléculas pequeñas (como los agentes con afinidad por los metales) o incorporación del fármaco a una macromolécula (p. ejem., los antimetabolitos).

![[Anteior]](../img/prevsec.gif)
![[Previo]](../img/parntsec.gif)
![[Siguiente]](../img/nextsec.gif)