VI. LA CONTRIBUCIÓN DE FARADAY A LA TEORÍA DE DISOCIACIÓN ELECTROLÍTICA
|
JOSÉ L. CÓRDOVA F
|
RESUMEN |
Se presentan los antecedentes científicos y filosóficos
que llevaron a Faraday a proponer la disociación electrolítica en los
términos del modelo de Boscovich. Se analizan y discuten someramente
los conceptos y experimentos científicos que permitieron a Svante Arrhenius
proponer, basado en la contribución de Faraday, la Teoría de Disociación
Electrolítica. El trabajo concluye mencionando que las investigaciones
de Faraday contribuyeron indirectamente al nacimiento de la fisicoquímica
como ciencia. |
EN 1850, nueve años antes de que naciera Arrhenius, 17 antes de
que muriera Faraday, el Journal of the Royal Agricultural Society of England
publicó dos artículos 13 ![[Nota 13]](../img/mcommnt.gif) que
describían el fenómeno del intercambio iónico en los terrenos de cultivo. En
uno de ellos se mencionaba que el cloruro de potasio podía ser retenido por
el terreno a pesar de las lluvias. El dispositivo experimental empleado para
comprobarlo era muy sencillo: una caja con tierra y perforaciones en el fondo.
Al analizar el líquido que salía de la caja después de verter solución de cloruro
de potasio, se encontró que contenía cloro pero nada de potasio; éste había
sido reemplazado por calcio y magnesio.
que
describían el fenómeno del intercambio iónico en los terrenos de cultivo. En
uno de ellos se mencionaba que el cloruro de potasio podía ser retenido por
el terreno a pesar de las lluvias. El dispositivo experimental empleado para
comprobarlo era muy sencillo: una caja con tierra y perforaciones en el fondo.
Al analizar el líquido que salía de la caja después de verter solución de cloruro
de potasio, se encontró que contenía cloro pero nada de potasio; éste había
sido reemplazado por calcio y magnesio.
Hoy día se representa lo anterior con:
La explicación, como toda aquella de carácter científico, es resultado de la colaboración de muchos científicos. En este trabajo se intenta ilustrar el papel que en ello tuvieron dos de ellos: Faraday y Arrhenius.
En tiempos de Faraday (1820) era del dominio científico la existencia de dos tipos de electricidad, originalmente llamados vítrea y resinosa. Las reacciones químicas se podían explicar entonces en términos de atracción y repulsión eléctricas. Así, los polos de un circuito atraían a distancia los componentes de una molécula, destruyéndola.
EL MODELO DE DISOCIACIÓN DE FARADAY
Michael Faraday notó experimentalmente que no era suficiente la cercanía de los polos eléctricos para producir una reacción química: hacía falta una descarga eléctrica. De una manera semejante había hallado que no se podía magnetizar agujas metálicas con polos eléctricos sino que se necesitaba una descarga. El experimento al que nos referimos consistió en colocar un papel filtro humedecido con solución de yoduro de potasio entre dos terminales cargadas eléctricamente. Cuando se libera yodo, el papel toma su característico color violeta, de forma que era fácil comprobar si había tenido lugar una reacción química. Sin embargo, repetimos, no bastaba la cercanía de los polos: debía producirse una descarga eléctrica para liberar el yodo.
A fin de interpretar el fenómeno, Faraday propuso que los componentes de las moléculas de yoduro de potasio emigraban en direcciones opuestas intercambiando continuamente de pareja hasta transformarse en especies químicas libres en los polos; la corriente eléctrica producía una perturbación a través de la solución química, que llevaba a la liberación de yodo.
Igual que su maestro H. Davy, Faraday conocía los trabajos de Gowin Knigth y del jesuita Roger Boscovich, cuyas obras capitales tienen títulos muy reveladores. La del primero es: Intento de demostrar que todos los fenómenos en la naturaleza pueden explicarse por dos principios activos simples: atracción y repulsión; y la de Boscovich: Teoría de filosofía natural elaborada segun la única ley de las fuerzas existentes en la naturaleza.
La unidad y convertibilidad de las fuerzas resultaba más consistente a las convicciones religiosas de Faraday, de manera que aceptó el modelo de Boscovich, del que se hablará a continuación.
EL MODELO ATÓMICO DE BOSCOVICH
Boscovich elaboró en 1758 un modelo atómico que combinaba las fuerzas de atracción y repulsión de manera que el patrón de fuerzas de un átomo boscovicheano sería como el que se muestra en la figura VI. 1.
La fuerza entre los átomos resultaba repulsiva a ciertas distancias y atractiva a otras. Nótese que, a distancias grandes, la fuerza era atractiva, de acuerdo con la ley de la gravitación de Newton; además, cada punto de intersección de la curva con el eje X representaba un punto de equilibrio, necesario para la existencia de compuestos químicos.
El modelo atómico de Boscovich partía de unas cuantas suposiciones y bastaba un solo tipo de partículas para explicar la gran diversidad de sustancias, y emplear, además, una sola curva de fuerza interatómica; con ello, las fuerzas químicas y gravitacionales se reducían a una sola. Como podrá suponerse, este modelo era puramente cualitativo; no se habían desarrollado instrumentos, ni experimentales ni matemáticos como para hacer predicciones cuantitativas.
![[FNT 13]](../img/136-94.gif)
Figura VI.1. Modelo atómico de Boscovich.
Ya se mencionó que la adhesión de Faraday al átomo de Boscovich tuvo raíces ideológicas. La doctrina de la unidad y convertibilidad de las fuerzas de la naturaleza (la Naturephilosophie alemana del siglo XIX) contradecía al materialismo inglés y ofrecía un lugar al mundo del espíritu. Faraday era un hombre muy religioso y es probable que optara por el modelo boscovicheano porque hallaba en él más coherencia con la creación de Dios, quien no dejaría a los átomos materiales en interacción caótica.
Según el modelo de Boscovich, los compuestos químicos eran agregados de átomos cuyas propiedades eran resultado de patrones de fuerzas de partículas puntuales. Si se permite el anacronismo, los átomos debían entenderse como interpenetraciones de campos de fuerza. Según esto, los átomos de Faraday no eran bolas de billar atractivas o repulsivas, sino centros de un complejo nudo de fuerzas.
Volviendo a la disociación del yoduro de potasio, la explicación que Faraday propuso fue la transmisión de una vibración gracias a la interacción de los átomos; en vista de que las fuerzas entre los átomos eran atractivas a unas distancias y repulsivas a otras, la perturbación se trasmitía en la solución hasta producir yodo libre.
La aportación de Faraday a la teoría de disociación electrolítica se puede resumir en los siguientes puntos:
1) El cambio químico se debe al paso de electricidad y no a la simple atracción eléctrica.
2) El cambio químico se da en los electrodos. Sólo scuando el ion yodo llega al electrodo se transforma en átomo de yodo.
3) El paso de la corriente eléctrica se debe a un intercambio de partículas de distinta carga: los iones.
Debe subrayarse que, para Faraday los iones eran producidos por la diferencia de potencial eléctrico. Como se verá más adelante, Arrhenius propuso que para formar iones bastaba con la disolución de una sal en agua, sin necesidad de polos eléctricos.
Aproximadamente en 1857, Rudolph Clausius postuló que en una solución existía un pequeño número de iones producidos por las colisiones azarosas de las moléculas. Puesto que en una solución, como en los gases, las partículas se mueven a muy diferentes velocidades, era posible que algunas tuvieran suficiente energía como para romper las moléculas del compuesto en una colisión. Según Johann Wilhelm Hittorf las partículas resultantes de estas colisiones transportaban la carga eléctrica en las soluciones, y eran los iones de Faraday.
Svante Arrhenius consideró las ideas anteriores al formular su teoría de disociación electrolítica que presentó en su tesis de doctorado (Universidad de Uppsala, 1884). No es ocioso decir que esta tesis recibió comentarios muy poco elogiosos por parte de sus profesores. Uno de ellos, Per Theodor Cleve (descubridor de los elementos holmio y tulio), afirmaba:
...no tiene ningún sentido decir que en una solución
de cloruro de potasio el cloro y el potasio están separados el uno del
otro... |
Arrhenius proponía que al disolver el cloruro de potasio en agua, los iones cloro y potasio se formaban sin necesidad de corriente eléctrica. Para entonces, la idea que prevalecía al respecto entre los químicos era la de Faraday: los iones se producen por el paso de la corriente eléctrica a través del electrolito; precisamente la palabra electrolito significa "destruido por la electricidad".
A pesar de las oposiciones, la tesis de Arrhenius fue aprobada pero no se le autorizó para ser profesor de la universidad.
Obviamente no era Suecia el lugar donde podía consolidarse la teoría, de modo que Arrhenius escribió a Rudolf Clausius, Lothar Meyer, Wilhelm Ostwald y Jacobus van't Hoff y les envió copias de su tesis. El entusiasmo e influencias de Ostwald y Van't Hoff ayudaron a difundir la teoría de Arrhenius, a pesar de la oposición de científicos de la talla de Mendeleyev, Kahlenberg y Lodge. El rechazo llegó a darse en términos tan acres como los de Lodge:
el senor Arrhenius es demasiado generoso en la
manipulación de datos imaginarios...de esta forma logra una enorme confusión
de la cual puede obtener sus llamadas deduciones teóricas. |
Sir Oliver Lodge sugirió, con 50 años de anticipación, la emisión solar de ondas de radio.
Ahora bien, entrando en materia, los experimentos hechos por Arrhenius para su tesis investigaban la conductividad de electrolitos empleando un depolarizador desarrollado poco antes. Este consistía en un conmutador manual que permitía cambiar el sentido de la corriente eléctrica varias veces por segundo, de manera que se evitaba la acumulación de cargas y, por consiguiente, la heterogeneidad de la solución. En otras palabras, con esta técnica Arrhenius logró, por primera vez, mediciones confiables de la conductividad de electrolitos.
Arrhenius trabajó además con soluciones muy diluidas (del orden de 5 x 10-4 equivalentes/litro), de manera que pudo llegar a conclusiones muy originales (aunque no totalmente fundamentadas). Por ejemplo:
1) Para soluciones muy diluidas, la conductividad específica de una solución salina es aproximadamente proporcional a su concentración en idénticas condiciones.
2) La conductividad de una solución diluida de dos o más sales es igual a la suma de las conductividades que tendrían las soluciones de cada sal a la misma concentración.
3) La conductividad de una solución es igual a la suma de las conductividades de soluto y solvente.
4) Si no se observan estas leyes esto se debe a que hay acción química entre soluto y solvente.
5) La resistencia eléctrica aumenta con el peso molecular del solvente.
Hoy día la quinta conclusión se considera incorrecta: la resistencia eléctrica aumenta con la constante dieléctrica del soluto (no con el peso molecular). Sin embargo, para las sustancias que Arrhenius empleó (agua, alcoholes, éteres), la constante dielétrica aumenta con la masa molar.
La importancia de la tesis de Arrhenius, con todo, no radica tanto en las mediciones experimentales cuanto en las ideas que al respecto comenzó a elaborar. Por ejemplo:
A estas partículas submoleculares las llamó iones, empleando el mismo término que Faraday. Como mencionamos antes, la proposición de una parte activa y otra inactiva ya la había hecho Clausius al afirmar que los choques entre las partículas podían romper las moléculas. Nótese, por otro lado, que la formulación de Arrhenius empleaba el concepto de equilibrio químico para explicar el comportamiento de las soluciones acuosas.
Continuemos con la tesis de Arrhenius:
...el calor de neutralización liberado por la transformación
de una base perfectamente activa y un ácido perfectamente activo es
sólo el calor de actividad del agua, donde el calor de actividad es
el calor empleado para transformar una sustancia del estado inactivo
al activo. |
El lector habrá observado que "perfectamente activo" significa completamente disociado, y que la reacción entre una base fuerte y un ácido fuerte es, simplemente:
Experimentalmente se encuentra que DH° es independiente de la naturaleza del ácido y la base, lo cual apoya fuertemente a la teoría de la disociación de Arrhenius. En efecto si DH° es constante es porque, posiblemente, siempre se tienen los mismos reactantes y los mismos productos. Considerando al ácido fuerte HA y la base fuerte BOH; si éstos, como afirma Arrhenius, están disociados completamente, dan lugar a:
y la reacción de neutralización es:
En resumen: la reacción de un ácido fuerte y una base fuerte es, simplemente, la reacción (1) y, en consecuencia, existen iones H + y OH- en solución sin necesidad de corriente eléctrica.
A fin de explicar las variaciones experimentales Arrhenius definió un "coeficiente de actividad" igual al cociente del número de iones realmente contenido en la solución y el número de iones que habría si el electrolito se transformara completamente en iones. Dicho de otra manera: mientras más fuerte fuese un ácido, base, etc., mayor sería su coeficiente de actividad y, de acuerdo con las ecuaciones de equilibrio químico, el coeficiente de actividad en la dilución infinita. Obviamente, las soluciones que empleó Arrhenius estaban muy diluidas (aproximadamente 5 x 10-4 equivalentes/litro).
Arrhenius también consideró la ley de migración iónica independiente de Kohlrausch:
donde L es la conductividad equivalente, m+,- es un coeficiente de proporcionalidad (llamado "movilidad iónica") y a es el "coeficiente de actividad" introducido por Arrhenius (hoy conocido como "grado de disociación").
Cuando Kohlrausch inició su investigación acerca de la capacidad de las soluciones para conducir corriente eléctrica, el tema era asunto de una gran controversia. Por ejemplo, se discutía la validez de la ley de Ohm para las soluciones, pues los resultados eran poco consistentes. Como se mencionó, la única corriente eléctrica disponible en el laboratorio era corriente continua, la cual produce polarización en los electrodos e influye en las mediciones, por Arrhenius se evitó ese problema utilizando un depolarizador.
También influían en las mediciones el tiempo empleado (pues se calentaba la solución), la intensidad de la corriente (afectaba la homogeneidad de la solución); sin embargo, a pesar de estas dificultades se pudo llegar a relaciones empíricas valiosas. Por ejemplo, la gráfica de conductividad equivalente de un electrolito (L) contra la raíz cuadrada de la concentración en equivalentes/litro (Öc), de la figura VI.2 es, para unas sustancias, una recta, mientras que para otras es una curva. Las sustancias que dan lugar a rectas fueron llamadas "electrolitos fuertes" por su gran reactividad química: HCl, KOH, KCl. Las otras como el CH3COOH, fueron nombradas "electrolitos débiles". Para los electrolitos fuertes, cuya gráfica es una recta, se pueden hacer extrapolaciones para dilución infinita; sin embargo, para los electrolitos débiles la extrapolación es poco precisa. El punto anterior es muy importante porque el comportamiento de una solución muy diluida es más predecible que el de una concentrada, pues las interacciones de las partículas del soluto son bajas.
![[FNT 14]](../img/136_99.gif)
Figura VI.2. Conductividad equivalente vs /c
En 1876 Kohlrausch pudo señalar que en soluciones muy diluidas los iones tienen
un movimiento independiente de su pareja. En una solución diluida cada especie
electroquímica tiene una conductividad propia. (cuadro VI.1).15 De
hecho Kohlrausch proponía que la conductividad de la solución era igual a la
suma de las conductividades de los componentes:
Se emplea L° para indicar conductividad a dilución infinita, pues en este caso la concentración de soluto es igual a cero.
El cuadro VI.1 muestra las conductividades de algunos electrolitos fuertes
a dilución infinita16 y
D representa la diferencia entre las conductividades
de dos soluciones con un ion común, por ejemplo:
pero por la proposición de Kohlrausch:
En el cuadro VI.1 se ve que el resultado loK+ - lo Li+ es independiente de las sales empleadas.
En el párrafo anterior se señaló la dificultad de determinar L, pues la curvatura de la línea hace poco confiable la extrapolación; sin embargo, la ley de Kohlrausch permite calcular L0 para el ácido acético.
Kohlrausch encontró cómo cuantificar una propiedad caracterísfica de las especies químicas que intervienen en el transporte de corriente en una solución. Esta propiedad es independiente del compuesto del cual procede y, evidentemente, no es afectada por el ion de carga opuesta. Sin embargo, para poder afirmar que los iones tienen existencia aun sin la corriente eléctrica hacían falta los trabajos de Raoult y las aportaciones teóricas de Clausius y Van't Hoff, que fueron sintetizados de manera elegante y completa por Arrhenius.
|
|
||
|
L0
|
||
|
Electrolito
|
Conductividad (m2 /ohm)
|
D = l0K+
- l0Li+
|
|
|
||
| KCl |
0.014986
|
|
| LiCl |
0.011503
|
3.483 X 10-3 |
|
|
||
| KClO4 |
0.014004
|
|
| LiClO4 |
0.010598
|
3.406 X 10-3 |
|
|
||
|
L0
|
D = l0Cl- - l0(NO3)- | |
| LiCl |
0.01503
|
|
| LiNO3 |
0.011010
|
4.93 x 10-4 |
|
|
||
| KCl |
0.014986
|
|
| KNO3 |
0.014498
|
4.90 x 10-4 |
|
|
||
| HCl |
0.042616
|
|
| HNO3 |
0.042130
|
4.83 x 10-4 |
|
|
||
LAS PROPIEDADES COLIGATIVAS Y LA TEORÍA DE ARRHENIUS
Por los trabajos de Clemens Heinrich von Babo (1847) y Adolf Wullner (1856) era un hecho conocido que la presión de vapor de un solvente disminuía al agregar un soluto y que dicha disminución era aproximadamente proporcional al peso del soluto. Desde tiempos de Fahrenheit se hacia uso de este efecto: él mismo lo empleó (en 1722) para definir uno de los puntos de referencia de su escala termométrica: la temperatura de fusión de una solución de sal y agua. Fahrenheit había notado también el aumento en la temperatura de ebullición por la presencia de un soluto.
Obviamente, en las observaciones de Fahrenheit, de Von Babo o de Wullner no se incluía a la masa molar del soluto, pues no se había desarrollado este concepto. Por otro lado, al experimentar con sustancias electrolíticas se obtenían resultados poco consistentes, como se explicará más adelante.
Francois Marie Raoult, por el contrario, empleó principalmente sustancias orgánicas (que generalmente son no electrolíticas) para investigar las propiedades de las soluciones (presión de vapor, temperaturas de ebullición y congelación). Encontró que los cambios en esas propiedades eran aproximadamente proporcionales al peso del soluto (1880). Ya con el desarrollo del concepto de masa molar, Raoult pudo hallar que la disminución del punto de congelación de cualquier solución de concentración 1 g en 100 g de agua, multiplicada por la masa molar, era una constante
|
|
||||||
|
0.001 molal
|
0.01 molal
|
0.1 molal
|
||||
|
Dtteor
|
Dtexp
|
Dtteor
|
Dtexp
|
Dtteor
|
Dtexp
|
|
|
|
||||||
| C12H22O11 | 0.00186 | 0.00186 | 0.0186 | 0.0186 | 0.186 | 0.186 |
| NaCl | 0.00186 | 0.00366 | 0.0186 | 0.0360 | 0.186 | 0.348 |
| K2SO4 | 0.00186 | 0.00528 | 0.0186 | 0.0501 | 0.186 | 0.432 |
| K3Fe(CN)6 | 0.00186 | 0.00710 | 0.0186 | 0.0626 | 0.186 | 0.530 |
|
|
||||||
Esta relación se podía aplicar también a la temperatura de ebullición y a la presión del vapor. Sin embargo, cuatro años más tarde Raoult reportó que esa relación fallaba con electrolitos, pero se aplicaba a los radicales que componían las sales como si éstos estuvieran separados en la solución.
Raoult empleó la ecuación 4 para determinar las masas molares de azúcares y alcoholes, sin embargo no investigó el origen de las desviaciones de las sales, ejemplificados en el cuadro VI. 2.
La ecuación 4 puede rearreglarse considerando que implica concentración de 1 g por 100 g de agua:
......................................................................= k2..............................(constante), (5)
.m2
y dividiendo la ecuación 4 entre la 5, obtenemos
......m1 .........k2
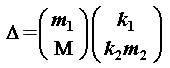
donde n es el número de moles de partículas de solutos y k3 una constante.
Es interesante observar que el valor Dtteor para las tres sales se aproxima a Dtexp cuando se multiplica respectivamente por 2, 3 y 4, respectivamente. Si el valor de Dt es función del número de partículas, como lo sugiere la ecuación 6, la conclusión será que hay 2, 3 y 4 veces el número teórico de partículas.
|
|
|||
|
molalidad =
|
0.001
|
0.01
|
0.1
|
|
|
|||
| NaCL |
1.97
|
1.94
|
1.87
|
| MgSO4 |
1.82
|
1.53
|
1.21
|
| K2SO4 |
2.84
|
2.69
|
2.32
|
| K3Fe(CN)6 |
3.82
|
3.36
|
2.85
|
|
|
|||
El mismo problema encontró Van't Hoff al investigar la presión osmótica de soluciones electrolíticas; para resolverlo ideó un coeficiente empírico i que corregía los resultados teóricos (Cuadro VI.3).
De hecho ambos términos i y a, están relacionados por la estequiometría de la reacción
En efecto, si consideramos la reacción 7 como reversible y en equilibrio químico, el número total de partículas (n ) en la solución es:
donde c° es la concentración molar inicial de la solución, y a el coeficiente de actividad de Arrhenius, es decir:
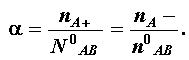
Los valores experimentales de i y a + 1 fueron tan aproximados que Arrhenius no titubeó en hablar de disociación a fin de explicar las anomalías en las propiedades coligativas. Para ello bastaba atribuir diferentes grados de disociación a las soluciones, lo que dependía de la naturaleza y concentración de las especies químicas.
Tomando como ejemplo una solución 1 M de NaCl, cuya temperatura experimental de congelación es -3.39° C, si se considera la reacción de disociación total:
habrá el doble de partículas en solución (una concentración 2 M) y una temperatura de congelación teórica de -3.72° C, mayor que el valor experimental. Arrhenius propuso que la reacción de disociación de la sal es reversible:
en ella el equilibrio químico entre las tres especies produce un número menor de partículas que el debido a una disociación total. Como puede verse en el cuadro VI.2, al disminuir la concentración de la solución de NaCL, el coeficiente empírico i se acerca a 2.
Sin embargo, ¿cómo admitir que aumenta el número de partículas en una sal sin una corriente eléctrica?, ¿cómo explicar que una partícula de Cl en la solución tenga propiedades físicas y químicas tan diferentes a las del cloro gaseoso? Tales fueron las principales objeciones a la teoría de Arrhenius la cual, sin embargo, pudo explicar:
a) las anomalías de la temperatura de congelación de electrolitos,
b) la diferencia entre electrolitos fuertes y débiles,
c) las anomalías en los valores de la presión osmótica de los electrolitos,
d) las anomalías en la temperatura de ebullición de electrolitos,
e) el valor constante del calor de neutralización de ácidos y bases fuertes.
f) la variación en la presión de vapor de las soluciones electrolíticas.
En una carta a Van't Hoff (30 de marzo de 1877), Arrhenius presentaba un razonamiento similar al presentado en este trabajo. La respuesta de Van't Hoff fue:
Su carta me ha aclarado considerablemente la constitución
de las soluciones. Puesto que los electrolitos se descomponen en sus
iones, el coeficiente i debe estar entre 1 y el número correspondiente
a la disociación. Con toda probabilidad los electrolitos se disocian
completamente en diluciones extremas. |
Sin embargo, a pesar de dar una interpretación elegante a una gran diversidad de fenómenos, la teoría de Arrhenius encontró muchas oposiciones. Como señalamos, Per Theodor Cleve, sinodal en el examen doctoral de Arrhenius, argumentaba que las cargas opuestas del cloro y del potasio impedían la separación de los iones. Arrhenius respondía que la atracción entre los iones era vencida por la de las moléculas del disolvente a los iones; si los iones no estaban hidratados (solvatados), no podían existir separados. Cleve preguntaba: ¿cómo podía existir cloro en una solución incolora e inofensiva de KCI siendo que el cloro es un gas amarillo verdoso y altamente oxidante?, ¿cómo podía existir potasio en tal solución sin liberar hidrógeno?
Hoy día tales preguntas parecen triviales (más por la familiaridad de los términos que por la comprensión de los conceptos); sin embargo, debe recordarse que en 1884 aún no se había descubierto el electrón. Sólo 16 años más tarde, con el concepto de electrón, pudo explicarse el origen de la carga eléctrica de los iones y la diferencia sustancial entre un átomo y su ion.
Si bien hay diferencias notables entre las teorías de disociación de Faraday y la de Arrhenius no puede dejar de mencionarse la influencia de Faraday. Esta va más allá del concepto de ion como partícula eléctrica: incluye el intento de unificación de fenómenos eléctricos y magnéticos, de fenómenos físicos y químicos.
El valor de ambas teorías no radica sólo en la belleza con que explicaron hechos aparentemente inconexos sino, sobre todo, porque permitieron plantear nuevos experimentos y leyes. La influencia de Faraday, a través de Arrhenius, llevó a la creación de una nueva disciplina: la fisicoquímica.
Concluyamos señalando que las objeciones y rechazos a la teoría de Arrhenius fueron explicados por él mismo cuando recibió el premio Nobel de química 1903.
La nueva teoría tuvo la mala suerte de que nadie
sabía dónde ubicarla: ni los químicos en la química, ni los físicos
en la física. Se tuvo que construir un puente entre ambas ciencias. |
Córdova, J. L. "Equivalente químico". Contactos, vol. II, núm. 6, 1985, pp. 44-52.
Córdova, J. L. "Teoría de disociación electrolítica. Un enfoque histórico". Contactos, vol. III, núm. 1, 1987, pp. 28-35.
Coulston, G. Dictionary of Scientific Biography. Ch. Scribner's Sons, Nueva York, 1976.
![[Inicio]](../img/begin.gif)
![[Anterior]](../img/prevsec.gif)

![[Siguiente]](../img/nextsec.gif)