V. CATÁLISIS HETEROGÉNEA
LA CATÁLISIS es esencialmente un fenómeno químico. La habilidad de una substancia para actuar como catalizador en un sistema específico depende de su naturaleza química. En catálisis heterogénea el fenómeno catalítico está relacionado con las propiedades químicas de la superficie del sólido que se ha elegido como catalizador, siendo por supuesto estas propiedades superficiales un reflejo de la química del sólido.
Una recopilación muy simple de catalizadores sólidos y de las reacciones que
éstos llevan a cabo condujo a Roginskii a proponer una relación entre propiedades
electrónicas y catalíticas.
|
|
|||
|
Tipos de sólidos
|
Reacciones
|
|
Catalizadores
|
|
|
|||
|
(conductores)
|
Hidrogenación deshidrogenación hidrólisis (oxidación)
|
[ | Fe, Ni, Pt, Pd, Ag, Rh, Ru |
|
(semiconductores) óxidos y sulfuros |
Oxidación Deshidrogenación desulfuración (hidrogenación)
|
[ |
NiO, ZnO, MnO2, Cr2, O3, Bi2O3-MoO3 WS2, MoS2 |
|
(aislantes) óxidos |
deshidratación
|
Al2O3, SiO2, MgO | |
|
ácidos
|
isomerización polimerización craqueo alkilación |
[ |
H3PO4, H2SO4 SiO2 - AL2O3 zeolitas |
|
|
|||
La tabla muestra que los metales de transición Fe, Ni, Pt, Pd, etc. son buenos catalizadores en reacciones que incluyen hidrógeno e hidrocarburos (hidrogenación, deshidrogenación, hidrogenólisis). Esto se debe a que esas moléculas interaccionan fácilmente con la superficie de esos metales.
Los óxidos (NiO, ZnO) son muy buenos catalizadores de oxidación debido a que
fácilmente interaccionan con el oxígeno y los hidrocarburos en su superficie.
Generalmente los óxidos son muy poco utilizados en hidrogenación porque durante
la reacción se reducen para dar metal (ZnO+H2 ![]() Zn
+ H2O). Los sulfuros se caracterizan por catalizar reacciones de moléculas conteniendo
azufre; si por ejemplo se usan óxidos para estas reacciones, éstos fácilmente
se sulfuran volviéndose inactivos.
Zn
+ H2O). Los sulfuros se caracterizan por catalizar reacciones de moléculas conteniendo
azufre; si por ejemplo se usan óxidos para estas reacciones, éstos fácilmente
se sulfuran volviéndose inactivos.
Existe una clase especial de óxidos como la alúmina (A12O3), la sílice (SiO2) y la magnesia (>MgO), los cuales no interaccionan mucho con el oxígeno y son, por lo tanto, malos catalizadores de oxidación. Sin embargo, estos óxidos interaccionan fácilmente con el agua y son muy buenos catalizadores de deshidratación.
Observamos entonces que existe cierta compatibilidad entre catalizador, reactivos
y productos. Para que el fenómeno catalítico ocurra, es necesaria una interacción
química entre el catalizador y el sistema reactivos-productos. Esta interacción
no debe modificar la naturaleza química del catalizador a excepción de su superficie.
Esto significa que la interacción entre el catalizador y el sistema reaccionante
se observa en la superficie del catalizador y no involucra el interior del sólido.
Este requerimiento nos lleva al concepto de adsorción.3![]()
La adsorción de moléculas de una fase fluida (gas o líquido) en la superficie
de un sólido está estrechamente ligada a la catálisis heterogénea. Todos los
sólidos tienen la propiedad de fijar (adsorber) en su superficie las moléculas,
átomos, o iones que se encuentren a su alrededor. Imaginemos una superficie.
Una superficie puede formarse por la ruptura de un cristal perteneciente a un
sólido covalente, como por ejemplo el diamante o cualquier metal. En el proceso
de ruptura del cristal, algunos enlaces covalentes entre átomos se rompen, lo
que origina que cada átomo en la superficie posea una o más valencias libres.
El número y tipo de estas valencias depende de la estructura del sólido y del
ángulo que haya sido utilizado para provocar la fractura. Cualquier átomo que
se localice en la superficie creada se encuentra en una posición poco usual,
el número de vecinos que poseía antes de la formación de la superficie ha disminuido
y experimenta un conjunto de fuerzas no balanceadas (Figura 7). Esta situación
conduce al fenómeno de energía libre superficial. Esta energía libre superficial
se podría comparar con la tensión superficial de los líquidos, sin embargo tiene
mayor fuerza debido a la mayor energía de cohesión de un sólido que de un líquido.
Si una molécula con afinidad hacia estas valencias libres se acerca lo suficiente,
se producirá un rearreglo electrónico con el sistema tal como se observa en
una reacción química. El resultado es la fijación de la molécula en la superficie
a través de una adsorción química o quimisorción.
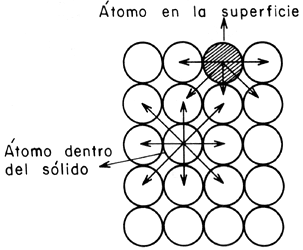
Figura 7. Representación de una superficie y balance de fuerzas en la superficie y el interior del sólido.
Algunas de las características de la quimisorción son:
1. Hay especificidad, sólo algunos sitios superficiales adsorben ciertas moléculas.
2. Hay una interacción de los estados electrónicos del adsorbato (gas) y del adsorbente (sólido), lo que se traduce en la formación de un verdadero enlace químico.
3. Como consecuencia de la reacción química superficial (rompimiento y formación de enlace) se desprende una cantidad elevada de calor.
4. La quimisorción requiere del suministro de una cierta cantidad de energía para iniciar el proceso (energía de activación). Proceso activado no espontáneo.
La otra forma de adsorción reconocida es la que ocurre por fuerzas del tipo Van der Waals, entre un átomo o una molécula y la superficie. En este caso no existe rearreglo electrónico en el sistema y sólo las fuerzas de atracción electrostáticas o atracciones dipolares son puestas en juego. A este tipo de interacción que ocurre sin modificación alguna de la molécula se le ha llamado adsorción física o menos frecuentemente, fisisorción.
Algunos criterios de distinción entre los dos fenómenos son mostrados en la
tabla 3.
|
|
||
|
Criterio de distinción
|
Quimisorción
|
Adsorción física
|
|
|
||
| Calor de adsorción (-D Hads) |
40-800 KJ / mol
|
8 - 20 KJ / mol
|
| Energía de activación |
Sí hay
|
No hay
|
| Temperatura |
Dependen de la Ea
|
Dependen del punto de ebullición
|
| Número de capas formadas |
Una
|
Más de una
|
|
|
||
En la quimisorción los nuevos enlaces formados en la superficie metálica son siempre en alguna medida polares debido a la diferencia de electronegatividad entre los átomos. Esto produce un cambio en el número de electrones de conducción en el sólido, lo cual puede ser fácilmente puesto en evidencia a través de medidas de conductividad eléctrica. En la fisisorción no ocurren tales cambios.
El proceso de adsorción en general es exotérmico. Siendo un proceso espontáneo,
DG es negativo:
Sin embargo, DS a su vez es negativo a causa de
que en la adsorción se produce un sistema más ordenado con pocos grados de libertad.
La sola posibilidad para que DS sea negativa es de
que DH sea mucho más negativa que DS
por lo que la adsorción es siempre exotérmica.
En la quimisorción el calor molar de adsorción es del orden de una reacción química 40-800 kJ/mol, en la fisisorción los calores son del orden del calor de licuefacción del gas.
Para que una reacción catalizada tenga lugar se requiere que la molécula sea primero quimisorbida en la superficie del sólido catalítico. Si dos moléculas van a reaccionar, al menos una de ellas debe estar quimisorbida.
Muchas moleculas se separan en el momento de la quimisorción. Por ejemplo la
molécula de hidrógeno se disocia en átomos de hidrógeno.
También la molécula de metano se disocia en la quimisorción:
Sin embargo algunas moléculas que tienen electrones p o un par de electrones
no apareados pueden quimisorberse sin disociarse, por ejemplo el etileno:
|
C2H4
+ 2M
|
||
|
|
|
|
|
|
|
M
|
M
|
|
donde M = átomo en la superficie de un sólido.
Otro ejemplo lo constituye el monóxido de carbono:
|
O
|
||
|
||
|
||
|
CO + 2M
|
||
|
/
|
\ | |
|
M
|
M | |
|
O
|
||
|
||
|
||
|
CO + M
|
||
|
||
|
||
|
M
|
||
En estos casos la molécula es adsorbida en forma asociativa. Una explicación más detallada acerca de la estructura de la molécula quimisorbida requiere una revisión de textos especializados. La adsorción es un fenómeno que se explica perfectamente utilizando un diagrama de energía potencial contra distancia a la superficie; tal diagrama se denomina de Lennard-Jones.
En la figura 8 se muesta la adsorción de hidrógeno en níquel. La abscisa representa
el punto de energía potencial cero. Así por ejemplo a una molécula muy alejada
de la superficie se le da una energía potencial de cero. Arriba de esta línea
se debe dar energía al sistema, abajo de esta línea el sistema está cediendo
energía al medio circundante.
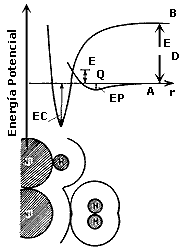
Figura 8. Curva de energía potencial para la adsorción de hidrógeno en níquel.
Examinaremos cómo cambia la energía potencial de la molécula de hidrógeno cuando se aproxima a la superficie del níquel, indicada por el eje de las ordenadas.
Al aproximarse la molécula de hidrógeno a la superficie se sigue el camino
A; a una cierta distancia las fuerzas de atracción y repulsión se minimizan
y la molécula se estabiliza con cierto potencial. En este momento ocurre la
adsorción física, y la cantidad de energía potencial cedida es el calor de adsorción
física (Ep). La distancia a la cual
la molécula se fija, rAF, es:
rAF
= rNi
+ rVDW(Ni)
+ rH
+ rVDW(H)
AF = .125
+ 0.08 + 0.35 + 0.08 = 0.32 nm. donde rVDW
= radio de Van der Waals.
Si se aproxima la molécula de H2 disociada a la superficie sigue
el camino B. Inicialmente hay una alta energía potencial (la energía suministrada
para la disociación = 434 kJ/mol). Conforme se acercan los dos átomos a la superficie
la energía potencial cae a un mínimo más profundo que el primero produciendo
el enlace de quimisorción a una distancia:
y liberando una energía aproximada de 125 kJ/mol. (Ec).
El punto más importante de este diagrama es que ambos caminos se cruzan a una distancia no muy arriba del cero de energía potencial (EQ). Así que para pasar una molécula de hidrógeno del estado de adsorción física al de quimisorción sólo se requiere suministrar una energía EQ que es la energía de activación de la quimisorción (mucho menor que la energía de disociación). Esta energía depende de la distancia mínima de la superficie, es decir del radio atómico de los átomos de la superficie y del adsorbato (lo que se adsorbe sobre la superficie). El punto de corte representa el estado de transición para la quimisorción.
De este esquema se deduce que la fisisorción es importante porque permite una quimisorción disociativa suministrando una energía menor que la necesaria para disociar la molécula ED (Figura 8).
Hemos visto que el fenómeno catalítico heterogéneo requiere de la adsorción química en la superficie del catalizador de al menos uno de los reactivos. Dado que la reacción se lleva a cabo en la superficie del catalizador, el conocimiento de la cantidad de moléculas adsorbidas en esta superficie reviste gran importancia. Debemos recordar que el sistema catalítico heterogéneo está constituido por un fluido que es una reserva de moléculas por transformar o ya transformadas y una superficie (catalizador). La concentración de reactivo adsorbido se relaciona por lo tanto con la concentración (presión) del reactivo en la fase gas (fluido). Para encontrar esta relación supongamos un sólido al cual se le suministra una cierta cantidad de gas (por ejemplo hidrógeno). Parte del gas se adsorberá en la superficie del sólido y parte quedará en la fase gas. Cuando la adsorción se ha completado y se alcanza el equilibrio, la relación entre la concentración de gas adsorbido y la presión del gas con la que está en equilibrio a temperatura constante se denomina isoterma de adsorción.
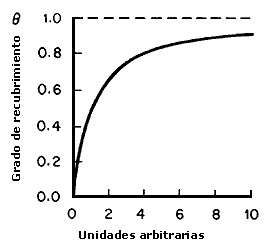
Figura 9. Isoterma de adsorción de Langmuir.
Siempre que el sólido sea no poroso y la temperatura se encuentre por
arriba del punto de ebullición del gas, la isoterma de adsorción tiene la forma
que se muestra en la figura 9. Imaginando el fenómeno, dos magnitudes pueden
ser fácilmente reconocidas: x, la cantidad adsorbida a una cierta presión P
de la fase fluida, y xmax que sería la cantidad máxima que la superficie
puede adsorber; definimos entonces la fracción de superficie recubierta como
q:
Para encontrar la relación matemática entre el grado de recubrimiento q y la
presión de equilibrio del gas imaginemos una superficie que consiste en n
"sitios" donde en cada "sitio" puede adsorberse una y sólo una molécula
del gas. El equilibrio que habíamos considerado anteriormente es de tipo
dinámico entre adsorción-desorción. El equilibrio puede representarse como:
|
adsorción
(ka)
|
|
|
A + S
|
|
|
desorción
(kd)
|
|
|
donde A
|
= reactivo |
|
S
|
= sitio en la superficie |
|
A-S
|
= reactivo adsorbido |
La velocidad de adsorción viene dada por la expresión:
y la velocidad de desorción por:
donde [A] es la concentración del reactivo A y que podemos sustituir por la presión PA al equilibrio, [S] representa la concentración de sitios vacíos y que podemos reemplazar por (1 - q), A-S es la concentración de sitios ocupados, la cual sustituimos por n q.
Cuando el equilibrio se alcanza, las velocidades de adsorción y desorción son
iguales, por lo que obtenemos:
que haciendo un poco de rearreglo nos queda:
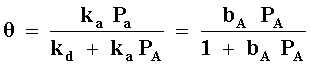
donde b = ka/kd se denomina coeficiente de adsorción de A en el sólido utilizado. Este término no es otra cosa que una constante de equilibrio cuya magnitud refleja la fuerza con que se adsorbe A, es decir, si b es muy grande, la molécula A se adsorbe fuertemente en la superficie.
La relación entre el grado de recubrimiento y la presión fue derivada por Irving Langmuir y se le conoce comúnmente como la isoterma de Langmuir.
El valor de b afecta a la forma de la isoterma de adsorción (Figura 10).
Mientras más grande sea el valor de esta constante, mayor será el grado de
recubrimiento a una presión de equilibrio dada. No todas las adsorciones obedecen
la isoterma de Langmuir. Esto se debe a muchas razones pero la más importante
es que se considera para su derivación el que todos los sitios en la superficie
son energéticamente equivalentes, lo cual rara vez se encuentra en
la práctica. Más aún, el calor de adsorción, en cual está cercanamente ligado
a la fuerza del enlace entre la especie adsorbida y la superficie, disminuye
al aumentar el grado de recubrimiento. De esta manera, otras isotermas han
sido derivadas para eliminar la suposición de la equivalencia energética de
los sitios; la isoterma de Temkin por ejemplo,
introduce las constantes k1 y k2
cuyos valores dependen del calor de adsorción inicial y supone una disminución
lineal del calor de adsorción con el grado de recubrimiento.
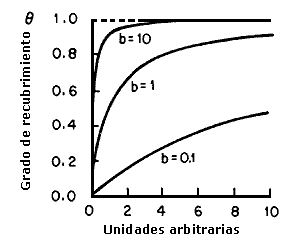
Figura 10. Variación de la isoterma de Langmuir con el valor de b.
La isoterma de Freundlich tiene la forma:
q = k P1/n
asumiendo en este caso una disminución logarítmica del calor de adsorción.
El conocimiento de la capacidad de adsorción de una sola capa de moléculas (monocapa) en un sólido no poroso puede ser fácilmente traducida en una medida del área superficial. La actividad (eficiencia) de un catalizador se expresa como la velocidad por unidad de área superficial (usualmente por m2) y de esta manera pueden compararse diferentes catalizadores. La isoterma de adsorción nos provee del número máximo de moléculas adsorbidas que pueden formar el recubrimiento correspondiente a una monocapa, es decir q = 1.0, luego entonces lo único que necesitamos es conocer el área que ocupa una molécula adsorbida para calcular el área total superficial:
Área total
sup = (número de
moléculas) X (área por molécula). Sin embargo, cuando se tiene un sólido poroso, la adsorción en multicapas
tiene lugar (Figura 11) y diferentes tipos de isotermas pueden observarse
(Figura 12). La información que puede obtenerse a través de estas isotermas
de adsorción física es: la superficie interna (poros), volumen de poro, distribución
de tamaño de poros, etc.
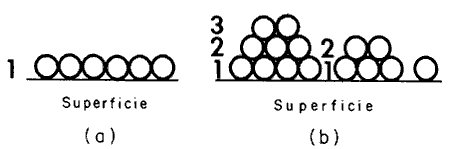
Figura 11. (a) Adsorción de una monocapa. (b) Adsorción en multicapas.
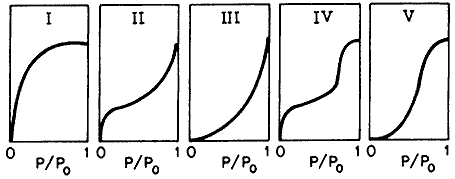
Figura 12. Tipos de isotermas de adsorción física.
El tipo I es de la forma de la isoterma de Langmuir y se observa para
sólidos microporosos4 ![]() incluyendo
zeolitas.
incluyendo
zeolitas.
El tipo II es el más común y aplicando la ecuación BET
(Brunauer, Emmett y Teller) que tiene la forma:
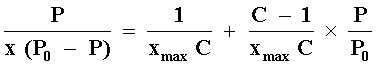
se puede obtener la capacidad de formación de una monocapa xmax. Po es la presión de vapor de saturación del gas que se adsorbe y C es una constante que involucra el calor de adsorción de la primera capa, con el calor liberado al formarse una segunda y subsecuentes capas. La formación de la monocapa se localiza en el punto B de la isoterma tipo II. Los tipos III y V son de poco interés pero el tipo IV es de importancia ya que presenta el fenómeno de histéresis, es decir la isoterma no sigue el mismo camino durante la desorción. La razón para esto es que la evaporación del gas condensado en los poros finos no ocurre tan fácilmente como la condensación, ya que una molécula que se evapora de una superficie curva (menisco) tiene mayor probabilidad de recondensar que una molécula que se evapora de una superficie plana. Este fenómeno permite de hecho determinar las distribuciones de tamaño de poro en sólidos porosos.
LA CINÉTICA DE REACCIONES HETEROGÉNEAS CATALIZADAS
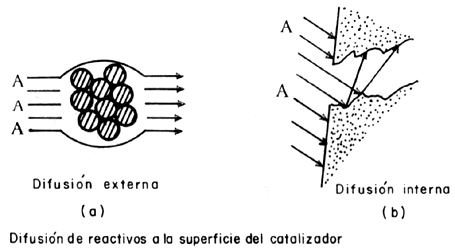
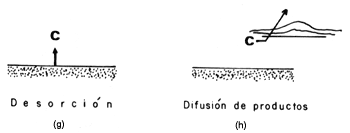
Cualquier reacción que tome lugar en una superficie comprende 5 pasos consecutivos
(Figura 13):
| 1) Difusión de reactivos a la superficie |
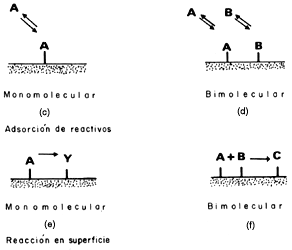
| 2) Adsorción de los reactivos |
| 3) Reacción en superficie |
| 4) Desorción de los productos |
| 5) Difusión de productos hacia la fase fluida |
Usualmente los pasos 1 y 5 son rápidos por lo tanto cualquiera de los pasos
2, 3 o 4 puede ser el paso limitante (el más lento) en cualquier reacción
heterogénea. Langmuir asumió que el paso 3, la reacción en superficie es el
paso lento del proceso, por lo que no es de extrañar que se utilice la isoterma
de Langmuir para estimar la concentración de especies adsorbidas.
|
|
|
Figura 13. Pasos involucrados en una reacción de superficie.
La determinación de parámetros cinéticos en una reacción catalizada es importante desde muchos puntos de vista. Por ejemplo, la determinación de los órdenes de reacción respecto a reactivos y productos es esencial para el establecimiento del mecanismo de la reacción cuyo conocimiento es indispensable para optimizar el catalizador. Asimismo, la información concerniente a los órdenes de reacción se utiliza para el diseño de reactores, tamaño y forma del lecho catalítico, etc. Otro parámetro cinético de gran importancia, la energía de activación, nos da información acerca de cómo la temperatura afectará la velocidad de la reacción.
El tipo de reacciones que más comúnmente encontramos en la realidad se ajustan
a los esquemas siguientes
| A + B |
|
C |
| X |
donde en el primer caso se forma un producto deseado (C) pero al mismo tiempo los mismos reactivos por un camino paralelo producen la especie (X) que puede ser no deseable. Este tipo de esquema de reacción se denomina de reacciones paralelas. En el segundo ejemplo, el producto (X) se obtiene por una reacción consecutiva del producto (C). Este tipo de reacción se denomina comúnmente reacción consecutiva.
En cualquiera de los dos casos es deseable conocer los parámetros cinéticos que rigen cada reacción. En ciertos casos, por ejemplo, el producto (X) puede permanecer en la superficie constituyendo un veneno para el catalizador, así, saber su velocidad de formación es vital.
En la cinética de reacciones heterogéneas se asume que la reacción en superficie es el paso limitante en la mayoría de las reacciones catalíticas heterogéneas, por lo que el conocimiento de la concentración de reactivo adsorbido en la superficie es un dato indispensable para derivar cualquier expresión cinética. La isoterma de Langmuir nos provee de tal información.
Supongamos primero una reacción donde el reactivo A adsorbido sin disociarse
se transforma en el producto C, el cual no se adsorbe. El reactivo A proviene
de la fase gas, se adsorbe, se transforma y el producto vuelve a la fase gas.
Una medida de la velocidad de reacción de A está dada por la velocidad de
desaparición de A en la fase gas y directamente relacionada con la concentración
de la especie adsorbida, es decir con el grado de recubrimiento de A en la
superficie del catalizador.
donde k es la constante de velocidad y qA el "recubrimiento" superficial del catalizador por las moléculas de A.
Este recubrimiento de A depende de la presión en la fase gaseosa de A, la dependencia más sencilla está dada por la ecuación de Langmuir:
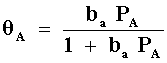
lo que nos da, substituyendo en la ecuación anterior
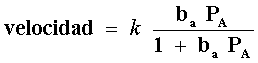
En esta ecuación, qA
![]() 1
entonces 1>> baPA
y nos queda v = kbaPa,
lo que significa un orden uno con relación a la presión de A.
Por el contrario, cuando qA
1
entonces 1>> baPA
y nos queda v = kbaPa,
lo que significa un orden uno con relación a la presión de A.
Por el contrario, cuando qA
![]() 0
se obtiene v = k lo que significa orden cero.
0
se obtiene v = k lo que significa orden cero.
Generalmente, las reacciones catalíticas son bimoleculares, es decir involucran dos reactivos, de esta manera tenemos para el proceso
A+B
que la velocidad de aparición del producto (C) está dada por la expresión
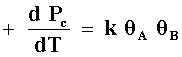
donde
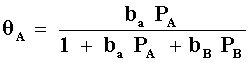 y
y 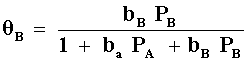
de aquí observamos que en el denominador aparecen los términos tanto de A como de B en ambas ecuaciones. Esto es debido a que en la adsorción de cada reactivo el otro está "compitiendo" por el mismo lugar. La ecuación de velocidad entonces quedará:
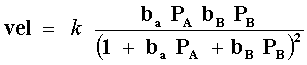
ecuación válida con las siguientes condiciones:
| a) De que las moléculas A y B se adsorban en el mismo sitio y sin disociarse (Figura 13 bis (a)). |
| b) De que el paso lento de la reacción sea la reacción entre las dos especies adsorbidas. |
| c) De que el producto C no se adsorba. |
Diferentes casos límite pueden darse, por ejemplo, si A y B, los dos, se
adsorben débilmente (el enlace entre ellos y la superficie no es muy fuerte),
es decir, bA y bB
son mucho menores que la unidad, la expresión se reduce a:
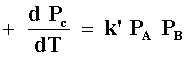
donde k' = kbAbB; la reacción es entonces de primer orden respecto a A y B y de orden total 2.
Otro caso límite es cuando A se adsorbe débilmente y B es fuertemente adsorbida, entonces bA <<1 + bB y la expresión de velocidad se reduce a (Figura 13 bis (a)):
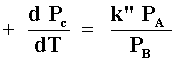
donde k" = ![]() ;
la reacción es primer orden respecto de A y menos primer orden respecto de
B. Este orden negativo tiene implicaciones importantes en el comportamiento
de la reacción cuando varían las presiones relativas de A yB en la fase gas,
se dice entonces que el reactivo B es un inhibidor.
;
la reacción es primer orden respecto de A y menos primer orden respecto de
B. Este orden negativo tiene implicaciones importantes en el comportamiento
de la reacción cuando varían las presiones relativas de A yB en la fase gas,
se dice entonces que el reactivo B es un inhibidor.
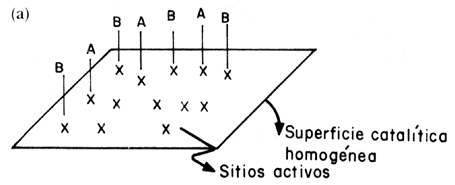
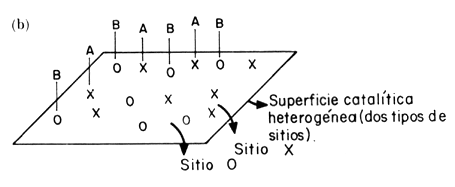
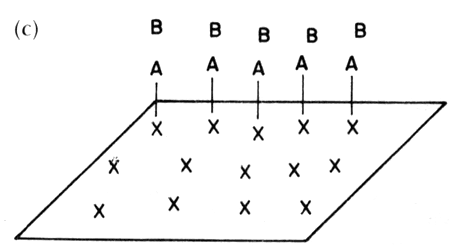
Figura 13bis. (a) Adsorción competitiva por los mismos sitios. Mecanismo Langmuir-Hinshelwood; (b) Adsorción en sitios diferentes (no competitiva). Mecanismo Langmuir-Hinshelwood; (c) Adsorción de un solo reactivo (A), el otro reacciona desde la fase gas. Mecanismo Eley-Rideal.
Otro caso de reacción bimolecular se presenta cuando los dos reactivos se
adsorben en sitios diferentes, es decir, no compiten por adsorberse en el
mismo sitio; entonces la expresión de velocidad toma la forma (Figura 13 bis
(b)):
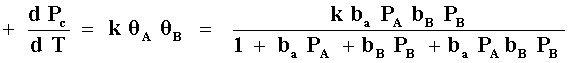
Otros casos incluyen la presencia de venenos, o adsorción del producto.
Hemos considerado hasta este momento que ambos reactivos deben estar adsorbidos
para que ocurra la reacción en superficie; este tipo de mecanismo se denomina
de Langmuir-Hinsherwood y puede ser representado como sigue (Figura 13 bis
(a) y (b)):
|
A(g) + S |
|
A - S
|
|
B(g) + S |
|
B - S
|
|
A - S + B - S |
|
C - S + S
|
|
C - S |
|
C(g) + S
|
donde S representa un sitio superficial y el índice (g) representa la fase gaseosa.
La primera ecuación representa el equilibrio de adsorción del reactivo A gaseoso sobre un sitio, la segunda es el equilibrio de adsorción del reactivo B. La tercera representa la reacción superficial entre A y B para formar C, finalmente la última es la desorción del producto C a la fase gas.
Existe otro tipo de mecanismo, por el cual, para que la reacción ocurra, sólo es necesaria la adsorción de uno de los reactivos y la transformación se efectúa al interaccionar esta molécula quimisorbida con las moléculas del otro reactivo que permanece en la fase gas (Figura 13 bis (c)).
Tal mecanismo se denomina de Rideal-Eley y la aplicación de la isoterma de Langmuir conduce a la expresión:
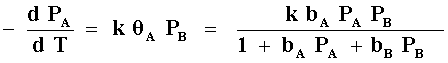
donde se asume que el reactivo B se adsorbe pero no reacciona.
El efecto de la temperatura en la velocidad de reacción se observará tanto
en la constante de velocidad como en el grado de recubrimiento. Para una reacción
unimolecular:
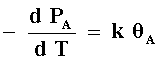
la constante k depende de la temperatura según la ley de Arrhenius:
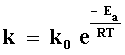 |
I
|
donde Ea es la energía de activación.
Tomando logaritmos naturales de la ecuación I y derivando respecto de la
temperatura obtenemos:
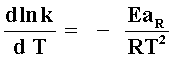 |
II
|
Para el grado de recubrimiento (qA), el efecto de la temperatura depende de la ecuación de Langmuir. Dos casos límite se observan:
a) adsorción débil (bAPA < 1), qA = bA PA y la velocidad se transforma en vR = kbAPA. Para el caso donde PA = constante, tenemos (k1 = kbA),
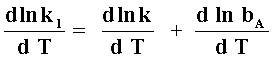
El primer término ![]() ,
relacionado con la ecuación II, nos proporciona la energía de activación
verdadera de la reacción. El segundo termino
,
relacionado con la ecuación II, nos proporciona la energía de activación
verdadera de la reacción. El segundo termino ![]() ,
está relacionado con la isócora de Van't Hoff:
,
está relacionado con la isócora de Van't Hoff:
introduce el calor molar de adsorción ![]() del
reactivo, luego entonces,
del
reactivo, luego entonces,
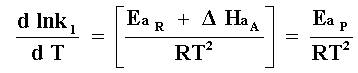
donde, EaR es la energía verdadera de activación, DHaA es el calor de adsorción
de A y Eap es la energía aparente de activación o energía de activación observada
experimentalmente. Por tanto,
ecuación que nos dice que la energía de activación observada no es la real. Como la adsorción es siempre exotérmica, DHa tiene un valor negativo, por tanto, - DHa tiene un valor positivo. La energía de activación verdadera EaR es entonces obtenida adicionando el calor de adsorción a la energía aparente de activación.
b) Adsorción fuerte, 1 << bAPA, por lo tanto
vR = k
de lo que se deduce que la energía de activación experimental es la energía verdadera de activación.
Para una reacción del tipo considerado, estudiado en un amplio rango de temperaturas, el comportamiento de la energía de activación se muestra en la figura 14.
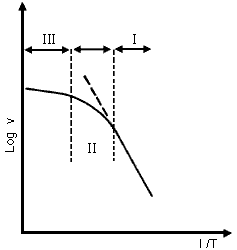
Figura 14. Diagrama ![]() de
Arrhenius para la reacción catalizada A à Productos.
de
Arrhenius para la reacción catalizada A à Productos.
|
|
|||
|
Caso
|
I
|
II
|
III
|
|
|
|||
| Recubrimiento q |
1
|
1>q>0
|
~0
|
| Orden de reacción n |
0
|
1>n>0
|
1
|
| Pendiente x 2.3 R,da |
|
-
|
|
|
|
|||
En la parte I se tiene un recubrimiento total de reactivo, obteniéndose un
orden cero y la energía de activación es la verdadera. En la fase II al aumentar
la temperatura el recubrimiento (qA)
disminuye, así como el orden; la velocidad de la reacción no sigue la ecuación
de Arrhenius. Finalmente, a temperaturas elevadas la superficie está casi
limpia (q ![]() 0),
el orden de reacción es 1 y la energía de activación no es la verdadera, sino
es sólo aparente e involucra el calor de adsorción del producto.
0),
el orden de reacción es 1 y la energía de activación no es la verdadera, sino
es sólo aparente e involucra el calor de adsorción del producto.